Project
Understanding the Detrimental Nature of Dislocations in Layered Cathode Active Materials II
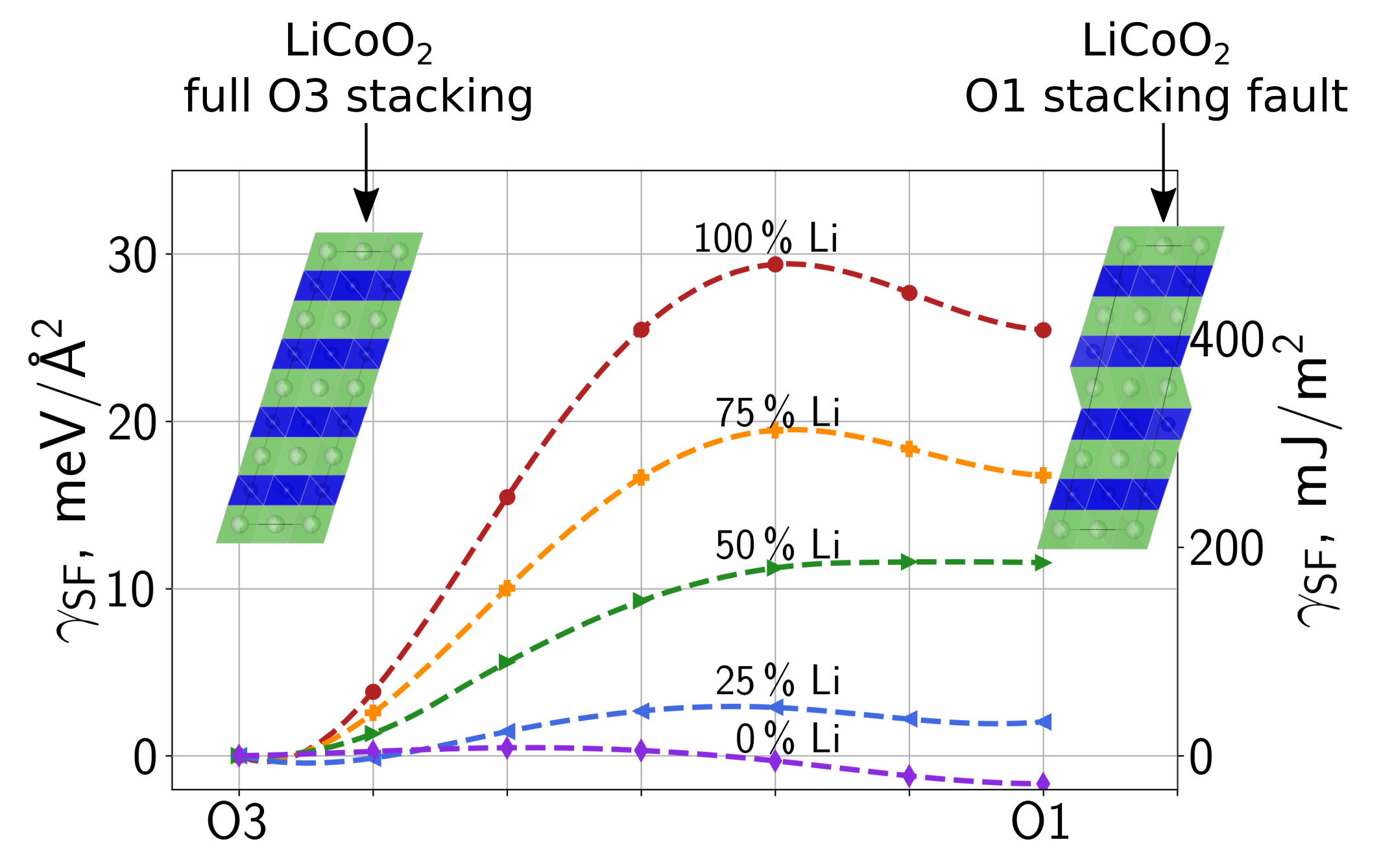 Figure 1: Gliding barriers for LiCoO₂ from a pure O3 stacking to a structure containing a O1 stacking fault for different lithium contents. The barriers have been obtained by performing solid-state nudged elastic band calculations within density function theory. The barriers and stacking fault energies are insightful to understand stacking sequence changes (i.e., phase transitions) as a function of the lithium content as well as the mechanical behaviour of the material.
Figure 1: Gliding barriers for LiCoO₂ from a pure O3 stacking to a structure containing a O1 stacking fault for different lithium contents. The barriers have been obtained by performing solid-state nudged elastic band calculations within density function theory. The barriers and stacking fault energies are insightful to understand stacking sequence changes (i.e., phase transitions) as a function of the lithium content as well as the mechanical behaviour of the material. Layered transition metal oxides are employed as cathode materials in high-voltage, rechargeable lithium ion batteries. The continuous delithiation and lithiation during charge and discharge, however, puts the material literally under stress: By changing the lithium content, phase transitions (i.e., certain lithium orderings or stacking sequence changes) as well as contraction and elongation of the lattice constants are induced. This couples to already present defects (e.g., Ni ions occupying the Li layer) and influences the generation of new defects such as oxygen vacancies or Ni migration, ultimately resulting in phases that resemble spinel or rock salt structures. These phases heavily deteriorate the properties of the material and are not well understood, yet. Therefore, we apply atomistic simulations to improve the understanding of the chemomechanical processes to derive optimization schemes for layered transition metal oxides.
Project Details
Project term
February 1, 2022–November 30, 2022
Affiliations
TU Darmstadt
Institute
Materials Modeling Division
Project Manager
Principal Investigator
Methods
We have mainly applied density functional theory (DFT) calculations using the Vienna ab initio simulation package (VASP) to optimize atomistic models and to calculate their electronic structure, total energies, forces and stresses. Initial models containing dislocations have been generated using the Babel package.
Results
The thermodynamics of layer gliding in LiCoO2 and LiNiO2 was investigated by performing solid-state nudged elastic band simulations by applying rigid gliding shift to the material. We find that both the gliding barrier (similar to the unstable stacking fault energy) as well as the stacking fault energy decrease with decreasing lithium content. The stacking fault energy becomes even negative towards 0% lithium content for both materials. In the case of LiNiO2, we also investigated how point defects influence the stacking fault energy by introducing an extra nickel in the lithium layer. At 0% Li content, 2% extra Ni has a similar effect on the barriers and stacking fault energies as a Li content of approximately 11% to 15%. Next, different models containing a dislocation dipole of screw dislocations for the two layered transition metal oxides were generated and treated within density functional theory. The excess energy of the dislocations are 0.83 and 0.48 eV/Å for LiCoO2 and LiNiO2, respectively. Additionally, also the fully delithiated materials were taken into account, resulting in excess energies of 0.30 and 0.40 eV/Å. Generally, full dislocations are able to split into two partial dislocations which reduces the energy of the system. The two partial dislocations then span a stacking fault. In our models, a splitting of the dislocations has also been observed after an advanced structural identification tool had been developed. Furthermore, we calculated defect binding energies between the dislocation and lithium and oxygen vacancies. For LiCoO2, both defects show overall negative binding energies. A comparison to elasticity theory (for which binding energies are obtained from strain and calculated defect dipole tensors) shows that inelastic contributions (only properly represented in density functional theory calculations) are responsible for the non-zero
binding energies. For LiNiO2, the binding energies follow a much more complicated pattern mostly alternating between positive and negative values. Again with the highest values are observed close to the dislcoation cores. Finally, as a first step towards understanding the generation of dislocations calculated surface energies of differently oriented surfaces. Based on these, a surface phase diagram was constructed and the particle morphology is predicted as a function of the chemical potentials for the atomic species. The different chemical potentials are motivated by different synthesis conditions (mainly temperature and oxygen partial pressure).
Discussion
The results of the layer gliding indicate that phase transitions with layer gliding are more likely to be observed towards low Li content. Moreover, at a Li content close to 0%, stacking changes are even thermodynamically favorable. In the context of the dislocations, the mechanical properties and elasticity theory can be used to understand one contribution to the difference in excess energies. Another contribution for LiNiO2 has been identified to relate to its Jahn-Teller distortions: We found that the Jahn-Teller distortions couple with to the high strain close the dislocation core. In this region, a reorientation of the Jahn-Teller distortions from its conventional zig-zag ordering to collinear regions is capable to take a portion of the strain, which lowers the excess energies considerably. For the delithiated material (NiO2) this is not possible because Jahn-Teller distortions become negligible due to the changed electronic structure of the Ni ion. The mostly negative binding energies of the vacancies in LiCoO2 indicate that an agglomeration of vacancies in the vicinity of the dislocation is favorable, potentially accelerating unwanted phase transitions. For LiNiO2, the more complicated patterns seem to relate to the locally differently oriented Jahn-Teller-distortions.
Additional Project Information
DFG classification: 406 Materials Science
Software: VASP, GULP
Cluster: Lichtenberg
Publications
M. Sadowski, L. Koch, K. Albe and S. Sicolo, “Planar gliding and vacancy condensation:
The role of dislocations in the chemomechanical degradation of layered transition metal
oxides”, Chemistry of Materials 2023 35 (2), 584-594 https://doi.org/10.1021/acs.chemmater.2c03069