Project
Full Optimization of Selected Configuration Interaction Wave Functions Using Quantum Monte Carlo Methods
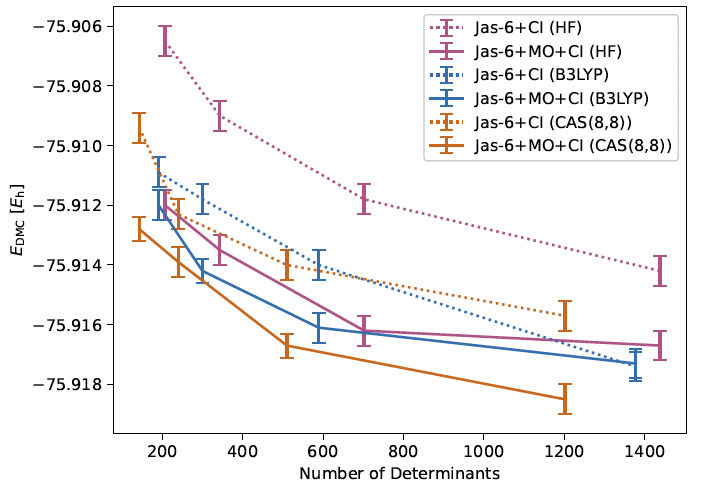 Figure 1: Zero time-step extrapolated DMC energies of truncated CIPSI-Jastrow wave
functions with different sets of initial orbitals as a function of the expansion
length. The data points are connected as guidance for the eyes.
Figure 1: Zero time-step extrapolated DMC energies of truncated CIPSI-Jastrow wave
functions with different sets of initial orbitals as a function of the expansion
length. The data points are connected as guidance for the eyes. The multi-reference DMC (MR-DMC) approach using CASSCF initial wave functions has been successfully applied to a range of transition metal dimers in terms of getting accurate dissociation energies. A considerable disadvantage of using CASSCF is based on its exponential growth within the active space, limiting actual calculations to about 20 electrons in 20 orbitals. Furthermore, within this approach, numerous configuration state functions (CSFs) that do not significantly contribute to the correlation energy are included in the wave functions. We, therefore, aim in this project at investigating truncated CIPSI-Jastrow wave functions, the CIPSI ansatz—a selected configuration interaction (sCI) technique—allowing to only choose energetically important determinants, thus generating more compact Slater-Jastrow wave functions. Some work has already been done with this approach employing HF initial orbitals. In this project, we intend to focus on the choice of initial orbitals in CIPSI-Jastrow wave functions. The use of quantum Monte Carlo (QMC) methods is justified not only because they are highly parallelizable but also because they scale favorably with the number of electrons. The variational (VMC) and diffusion (DMC) quantum Monte Carlo approaches are the most widely spread stochastic methods applied in chemistry and physics to determine the properties of systems when an accuracy beyond mean-field theory is required. In this project, we studied the 1Σ+g ground state of C2 by means of truncated CIPSIJastrow wave functions using VMC and DMC. The original CIPSI wave functions—with different initial orbitals—were truncated at several cutoff values, corresponding to the absolute values of the CI coefficients. The Jastrow, MO, and CI parameters of the obtained wave functions were either partially or fully optimized within the VMC framework with respect to the variational energy. In a next step, DMC calculations were performed for each wave function.
Project Details
Project term
May 15, 2022–November 14, 2022
Affiliations
RWTH Aachen University
Institute
Institute of Physical Chemistry
Project Manager
Principal Investigator
Methods
Using initial HF orbitals, we computed the bond dissociation energy of C2 for the fully optimized CIPSI-Jastrow wave functions (for several C2 thresholds) at DMC level and found that accurate dissociation energies can be obtained when the truncation scheme for the carbon atom is carefully selected. The best results were obtained when using the same wave function norm as threshold for both the carbon atom and the dimer,justified by the same amount of correlation being lost for both species. Furthermore, we showed that the CI picture is significantly changed if the molecular orbitals and the CI coefficients are re-optimized in the presence of the Jastrow correlation function. This is fundamental since it reveals the importance of choosing configurations in the presence of the Jastrow factor. We are, therefore, currently working on developing an sCI algorithm within QMC that chooses the CSFs with the Jastrow factor present. In the second part of the project, different sets of initial orbitals, namely Kohn-Sham (KS)-B3LYP, KS-PBE0, and CASSCF were employed for the generation of the CIPSI wave functions and the resulting VMC optimized truncated CIPSI-Jastrow wave functions were compared with their counterparts using HF initial orbitals. Figure 1, expemplarily, displays the DMC energies of C2 for different ans¨atze as a function of the number of determinants. The KS orbitals, using the B3LYP exchange correlation functional, provided superior results compared to the KS-PBE0 initial orbitals. The data for PBE0 are not shown in the figure for clarity reasons.
Results
Figure 1 exhibits, first of all, that the expansion size is reduced for a given threshold when using KS orbitals—compared to HF orbitals—and even more so when CASSCF orbitals are employed. The shorter expansions—for a given cutoff—imply that the (KS and CASSCF) orbitals themselves—compared to HF—already account for some amount of correlation. At the Jas+CI optimization level, (mostly) lower DMC energies are obtained when using initial KS orbitals, the difference in energy being much more prominent for the HF initial orbitals than for the CASSCF counterparts. It should be noted that, for the largest expansion, no improvement is achieved for the initial KS orbitals when the molecular orbitals are re-optimized within VMC. This may imply that one might be able to dispense with the MO optimization, which is rather costly especially for larger systems. Finally, at the Jas+MO+CI optimization level, the CIPSI-Jastrow wave functions with CASSCF initial orbitals yield the lowest DMC energies for each cutoff. However, one should keep in mind that the choice of the active space (in CASSCF) has to be performed manually which constitutes a drawback of using CASSCF initial orbitals.
Additional Project Information
DFG classification: 303-02
Cluster: CLAIX
Publications
Jil Ludovicy, Robin Dahl, and Arne Lüchow, Toward Compact Selected Configuration Interaction Wave Functions with Quantum Monte Carlo—A Case Study of C2, J. Chem. Theory Comput. 2023, under review.