Project
True ab initio spin dynamics and thermodynamics from massively parallel constrained supercells in spintronic and caloric materials
 Figure 1: Magnetically constrained DFT calculations at different atom-scale magnetic orientations, used to describe thermal excitations and dynamics. The magnetic system is fully ordered at absolute zero temperature (a), while it becomes disordered as the temperature raises (b,c).
Figure 1: Magnetically constrained DFT calculations at different atom-scale magnetic orientations, used to describe thermal excitations and dynamics. The magnetic system is fully ordered at absolute zero temperature (a), while it becomes disordered as the temperature raises (b,c). 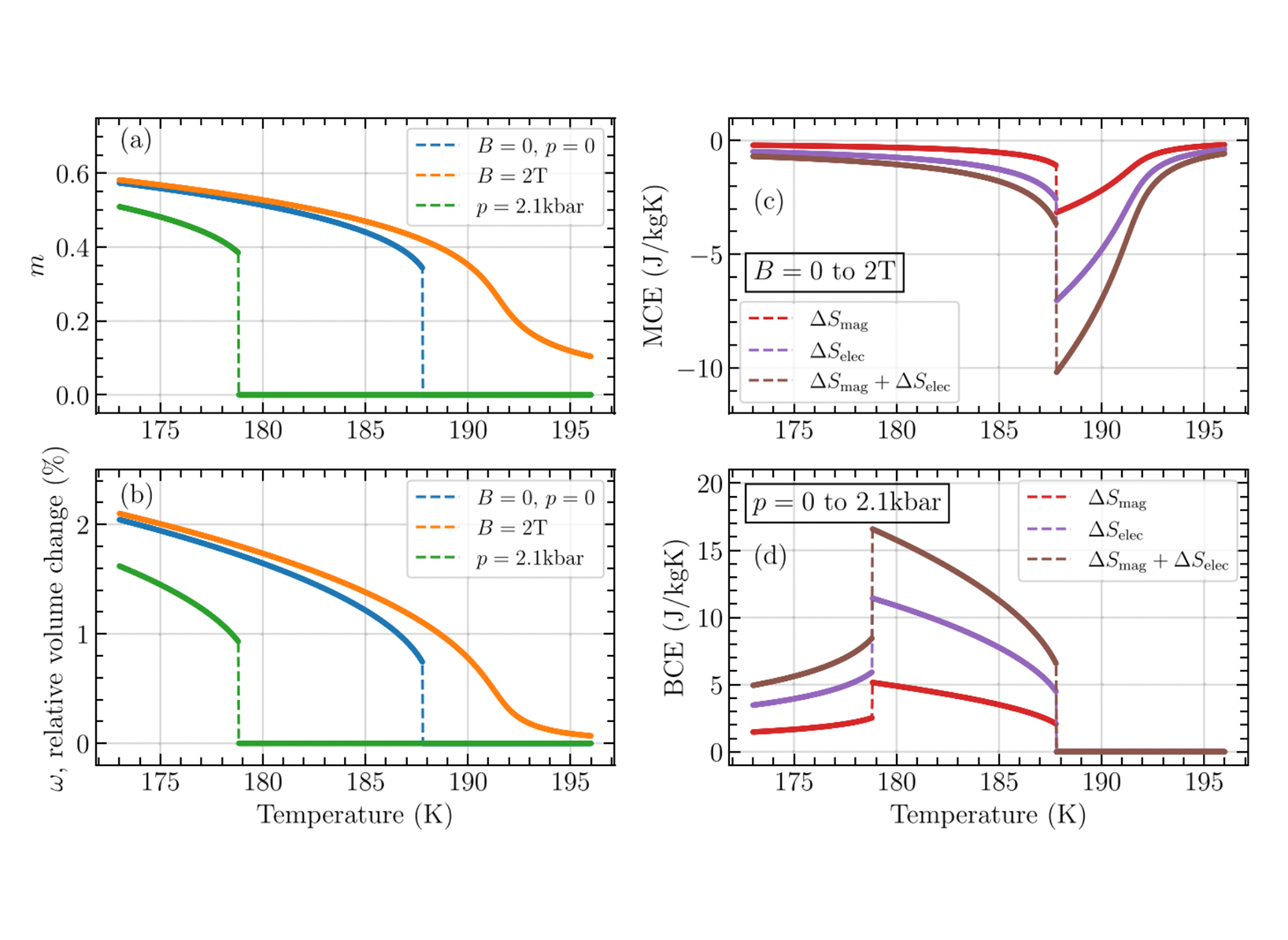 Figure 2: Temperature dependence of (a) reduced magnetisation and (b) relative volume change computed for a La-Fe-Si system with (c,d) giant cooling effects.
Figure 2: Temperature dependence of (a) reduced magnetisation and (b) relative volume change computed for a La-Fe-Si system with (c,d) giant cooling effects. Many modern solid-state technologies that greatly impact our society are based on magnetic materials. These include devices exploiting spintronic phenomena, as for neuromorphic computing and novel nonvolatile memories, devices for mobility, ground transportation and energy generation, as well as the creation of new types of solid-state refrigeration. The latter is the so-called caloric cooling, which promises to be an energy efficient and environmentally friendly alternative to urgently combat the climate change. In order to make key progress in the understanding and development of these technologies one must study the fundamental element underlying the exceptional properties of magnetic materials: the atom-scale magnetic polarizations known as magnetic moments or spins. These magnetic moments have magnetic orientations that can form different ordered patterns or magnetic states. Their dynamics and fluctuations, as well as transitions between them, underline the functionality of both spintronic and caloric applications. However, their investigation and prediction from first-principles, i.e., using fundamental laws of nature only, is a great challenge merely accessible by a few research groups in the world. One of the few computational approaches able to perform such tasks is based on the combination of Density Functional Theory (DFT) together with constraints of the magnetic moments to specific orientations. This project focused on the development and application of computational tools for the study, understanding, and prediction of spintronic and caloric materials by means of magnetically constrained DFT calculations.
Project Details
Project term
August 1, 2022–July 31, 2023
Affiliations
Forschungszentrum Jülich
University of Barcelona
Institute
Peter Grünberg Institut
Project Manager
Principal Investigator
Methods
Density functional theory (DFT) forms the basis of most materials modelling that is based on first-principles calculations. It is the worldwide most successful approach for the computation of the electronic structure of materials, requiring the use of supercomputers to describe crystals containing multiple sublattices. However, standard DFT is typically confined to the absolute zero temperature and to specific magnetic moment orientations that minimize the magnet’s total energy. It is, therefore, usually far from providing a robust thermodynamic description of both caloric cooling and spin dynamics. DFT can be extended to include constraints of the orientations of the magnetic moments in order to explore thermal excitations and transient magnetic states. The underlying tenet behind this picture is to assume that the magnetic moment orientations evolve in a time-scale that is much larger than that one for the fast moving electrons. This framework allows the implementation of two different DFT-based first principles tools:
(1) The Disordered Local Moment (DLM) theory, one of the few finite-temperature approaches of magnetism that exists, which describes thermal excitations of the local moments by employing the coherent potential approximation to simulate different states of magnetic disorder at finite-temperature.
(2) The computation of magnetictorques in transient magnetic states for their input in the Landau–Lifshitz–Gilbert
equation for the description of spin dynamics in magnetic systems. All the DFT codes used in this project are based on the Korringa-Kohn-Rostoker (KKR) electronic structure method. While one of them has been already designed to directly implement the DLM picture (MARMOT package), we recently implemented constraining magnetic fields in another of our DFT codes to perform spin dynamics and thermodynamics from massively parallel supercell calculations
(KKRnano package).
Results
One of the central objectives of our project was the investigation of the dependence of the energy functional of a magnetic material at finite temperature on different key parameters with the aim to uncover universal behaviours with important repercussions on the material’s potential as a caloric refrigerant. Our computational tool implementing the DLM-DFT theory is able to compute internal energy coefficients that directly determine the nature of a functional transition between different magnetic states that generate caloric cooling. Our calculations can be performed to study the effect of the crystal structure, material’s magnetization, and supercell size (when using KKRnano) on the refrigerant transition in order to engineer optimal materials. These investigations have focused on two different material families: (a) antiperovskite systems Mn3AN, which is a famous magnetic materials class that shows giant caloric cooling that can be modified by chemical doping of element ”A”, and (b) La(FexSi1−x)13 compounds, which are well-known for their giant caloric responses with low hysteresis. Work is in progress regarding the investigation of Mn3AN’s dependence of internal energy coefficients on supercell sizes using KKRnano aiming at predicting accurate free energies of materials requiring the construction of smaller supercells only. On the other hand, our calculations on La(FexSi1−x)13 have produced important results demonstrating that the origin of its refrigerant transition is due to a large coupling of magnetism with the volume. This represents the first ever given explanation of such an important caloric transition fully from first-principles, which has been already published.
Discussion
Although work regarding the universal behaviour of the internal energy coefficients with respect to the unit cell size is still in progress, our current results show a good comparison with those obtained using the coherent potential approximation for Mn3AN, which indicates that our proposed approach based on supercell calculations is feasible in these systems. Other supercell calculations carried out for La(FexSi1−x)13 materials present, however, some differences. This calls for further investigations in this material since our supercell approach can be more accurate for the modellization of the electronic structure. On the other hand, we have developed a python class to efficiently interface KKRnano with the Landau–Lifshitz–Gilbert equation. Several tests to evaluate its performance and preliminary results regarding the calculation of first principles spin dynamics in Mn3AN and other materials of interest are available.
Additional Project Information
DFG classification: 307-02 Theoretical Condensed Matter Physics
Software: MARMOT, KKRnano
Cluster: CLAIX
Publications
E. Mendive Tapia, C. E. Patrick, T. Hickel, J. Neugebauer, and J. B. Staunton,
”Quantification of electronic and magnetoelastic mechanisms of first-order magnetic
phase transitions from first principles: application to caloric effects in La(FexSi1−x)13”,
Journal of Physics: Energy 5, 034004 (2023)
Nicolas Essing, ”Ab-Initio Spin Dynamics using the KKRnano method”, RWTH
Aachen University, 2022, master thesis, (upon request, contact s.bluegel@fz-juelich.de)