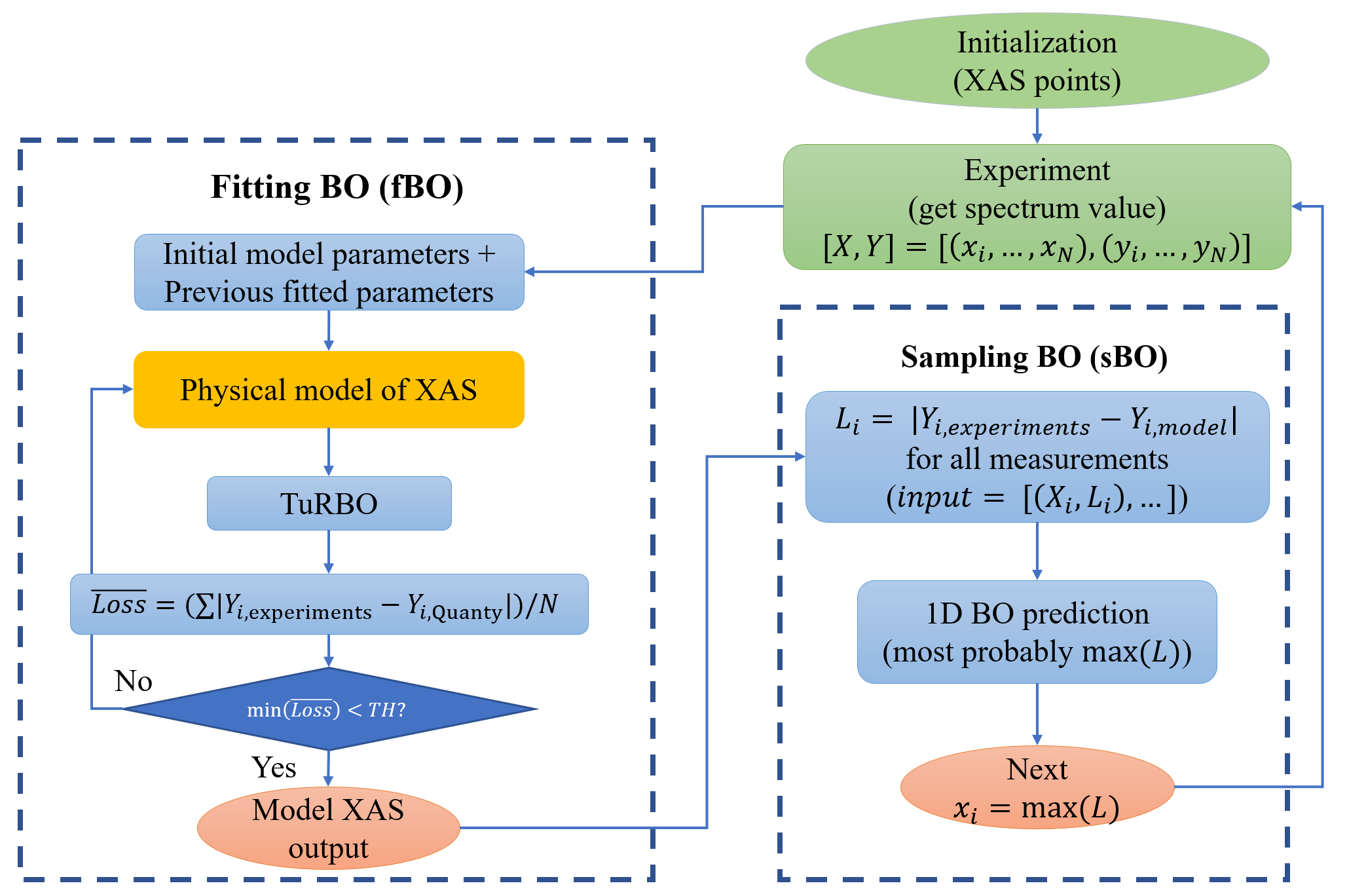
 Figure 1: Workflow
Figure 1: Workflow 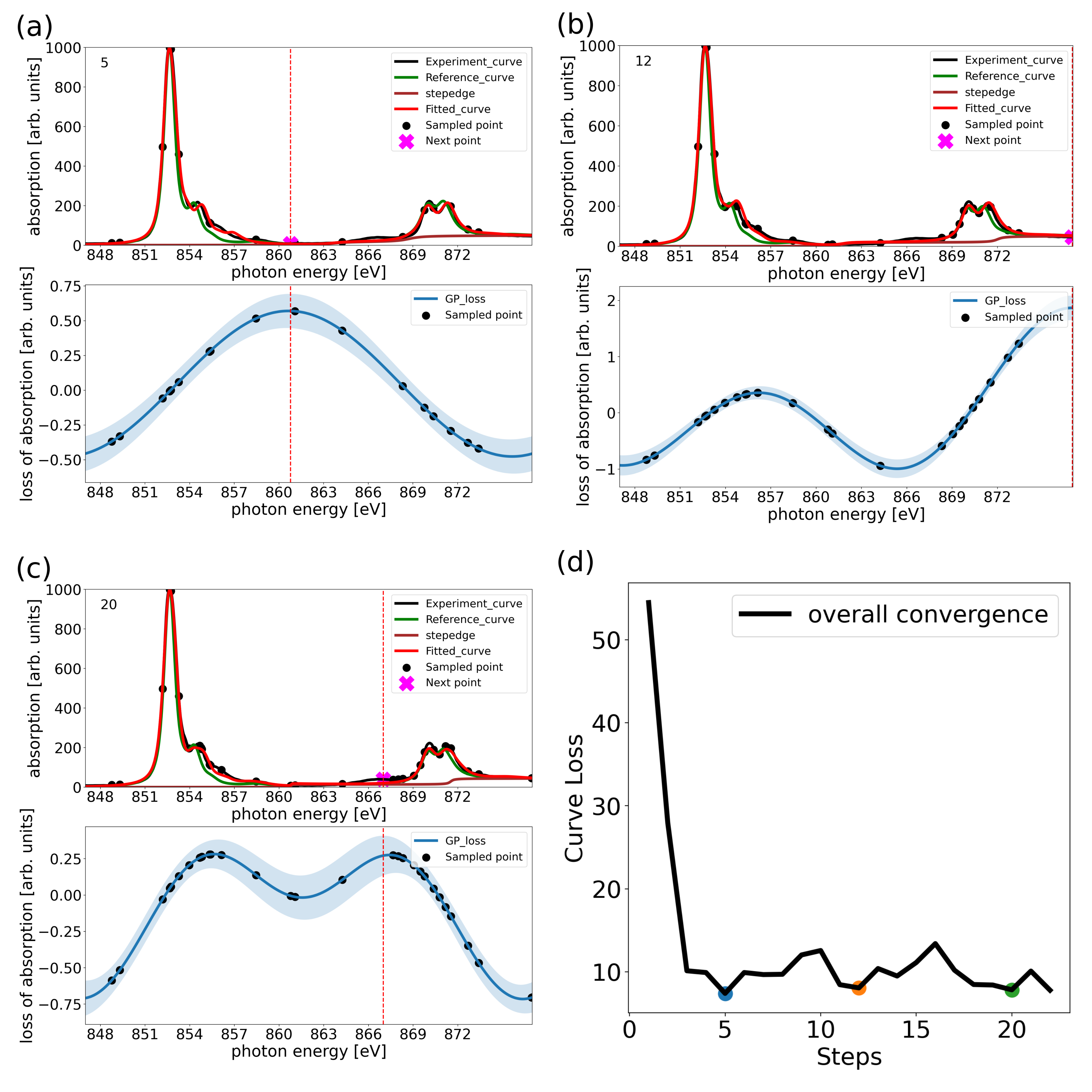 Figure 2: ABO fitting and sampling of experimental NiO XAS curve with automatic background subtraction
Figure 2: ABO fitting and sampling of experimental NiO XAS curve with automatic background subtraction X-ray Absorption Spectroscopy (XAS) is a pivotal technique in material research, requiring numerous sampling points for detailed analysis and complex atomic Hamiltonian-based data interpretation. Various methodologies such as Density Functional Theory (DFT), semi-empirical multiplet models, and a combination of DFT and Dynamical Mean-Field Theory (DFT+DMFT) are employed for XAS interpretation, each with its own advantages and limitations. While DFT captures the crystalline environment effectively, it struggles with correlation effects, which DFT+DMFT addresses but at a high computational cost. On the other hand, semi-empirical methods such as multiplet model Hamiltonian offer a transparent mechanistic understanding of XAS but can lack in capturing certain interactionseffectively. How to combine the merits of both side is of vital importance. This work introduces the Automated Bayesian Optimization (ABO) algorithm for efficient parameter fitting of physical model and experimental sampling based on active learning, significantly reducing the required number of sampling points while ensuring precise XAS analysis. Using NiO as a test case, the ABO algorithm demonstrated its capability to construct complex models with a limited data set, distinguishing between different physical processes and applying the methodology to real experimental data. This approach presents a valuable solution for time-efficient and accurate XAS measurements and analysis, paving the way for more efficient material property investigations.
Project Details
Project term
January 1, 2023–January 31, 2023
Affiliations
TU Darmstadt
Institute
Theory of Magnetic Materials
Project Manager
Principal Investigator
Methods
The physical model of Ni2+ in the nickel oxide L2,3 edge XAS was constructed using Quanty software, employing model Hamiltonians based on crystal field multiplets (CFM) and charge transfer multiplets (CTM). The evaluation procedure employs a Lanczos-based Green’s function method for both ground and excited states and applies Gaussian and Lorentzian broadening methods to bring the results into close agreement with experimental observations. The ABO algorithm is similar to Generative Adversarial Networks (GANs) in that it integrates two main components: fitting Bayesian Optimization (fBO) to minimize the variance in the XAS, and sampling Bayesian Optimization (sBO) to sample the points that could lead to the failure of the current fBO model. Each component of Bayesian optimization involves the use of a Gaussian Process (GP) to fit the XAS intensity, which requires normalization and optimization of the kernel function. At the same time, a tailored acquisition function tuning strategy is required depending on the model and data situation. To cope with the challenges of high dimensionality and the possibility of falling into local optima, the trust region Bayesian optimization (TuRBO) method is also employed. The method advocates adaptive tuning of the trust region, aiming to maintain the accuracy of the local surrogate model while facilitating the global search, and the parallelization of the trust region is considered to be an effective strategy for locating the global optimum.
Results
In the case of nickel oxide (NiO), the algorithm demonstrates that fewer than 30 sampling points are sufficient to reconstruct the entire XAS spectrum and align it with the associated crystal field or charge transfer model, which can be automatically selected during the sampling process based on intuitive hypothesis learning. When applied to experimental XAS spectra, reasonable XAS and plausible atomic model parameters can be obtained with fewer than 80 sampling points. This efficiency not only simplifies the analysis process, but also ensures the reliability of the atomic model parameters.
Discussion
The ABO algorithm provides reasonable physical parameters consistent with the results while sampling efficiently by automatically fitting multi-body model Hamiltonians and using them as a priori data for the Active Learning sampling method. These results highlight the potential of the ABO algorithm as a physically-driven AL method for efficient sampling and accurate analysis in XAS, and provide a solid foundation for future applications in this area.
Additional Project Information
DFG classification: 406 Materials Science
Software: VASP
Cluster: Lichtenberg
Publications
Zhang, Y.; Xie, R.; Long, T. et al. Autonomous atomic Hamiltonian construction and active sampling of X-ray absorption spectroscopy by adversarial Bayesian optimization, npj Comput. Mater., 9, 46 (2023). https://doi.org/10.1038/s41524-023-00994-w
Shen, C.; Li, T.; Zhang, Y.; Xie, R. et al. Accelerated Screening of Ternary Chalcogenides for Potential Photovoltaic Applications, J. Am. Chem. Soc., 145, 40, 21925–21936 (2023). https://doi.org/10.1021/jacs.3c06207
Hu, K.; Xie, R.; Shen, C. et al. High-throughput design of Co-based magnetic Heusler compounds, Acta Mater., 259, 119255 (2023). http://dx.doi.org/10.2139/ssrn.4400799