Project
Molecular Dynamics Simulations of Anchor Peptides−Surface Interactions (Pepsursim)
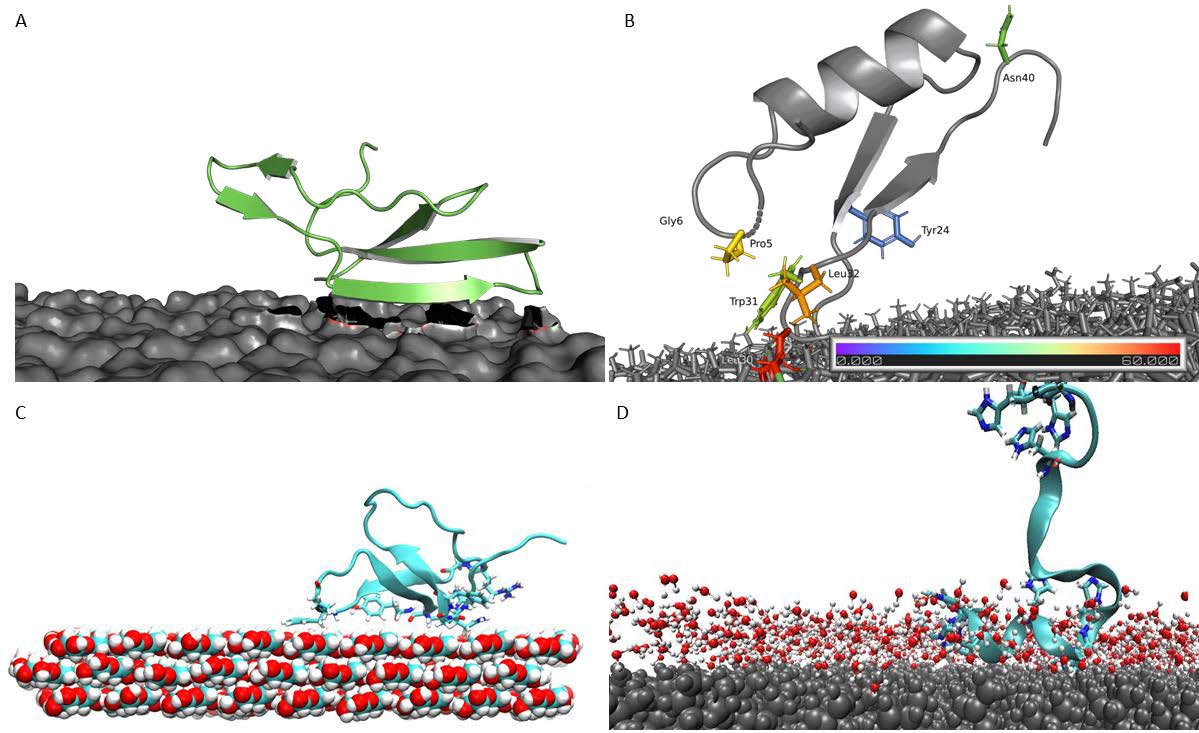 Figure 1: Most common binding poses of APs on respective surfaces. A: LCI WT on the PLA surface. B: CgDef WT on PLA. C: TA2 on a hydrophobic cellulose surface. D: MacHis on PP.
Figure 1: Most common binding poses of APs on respective surfaces. A: LCI WT on the PLA surface. B: CgDef WT on PLA. C: TA2 on a hydrophobic cellulose surface. D: MacHis on PP. 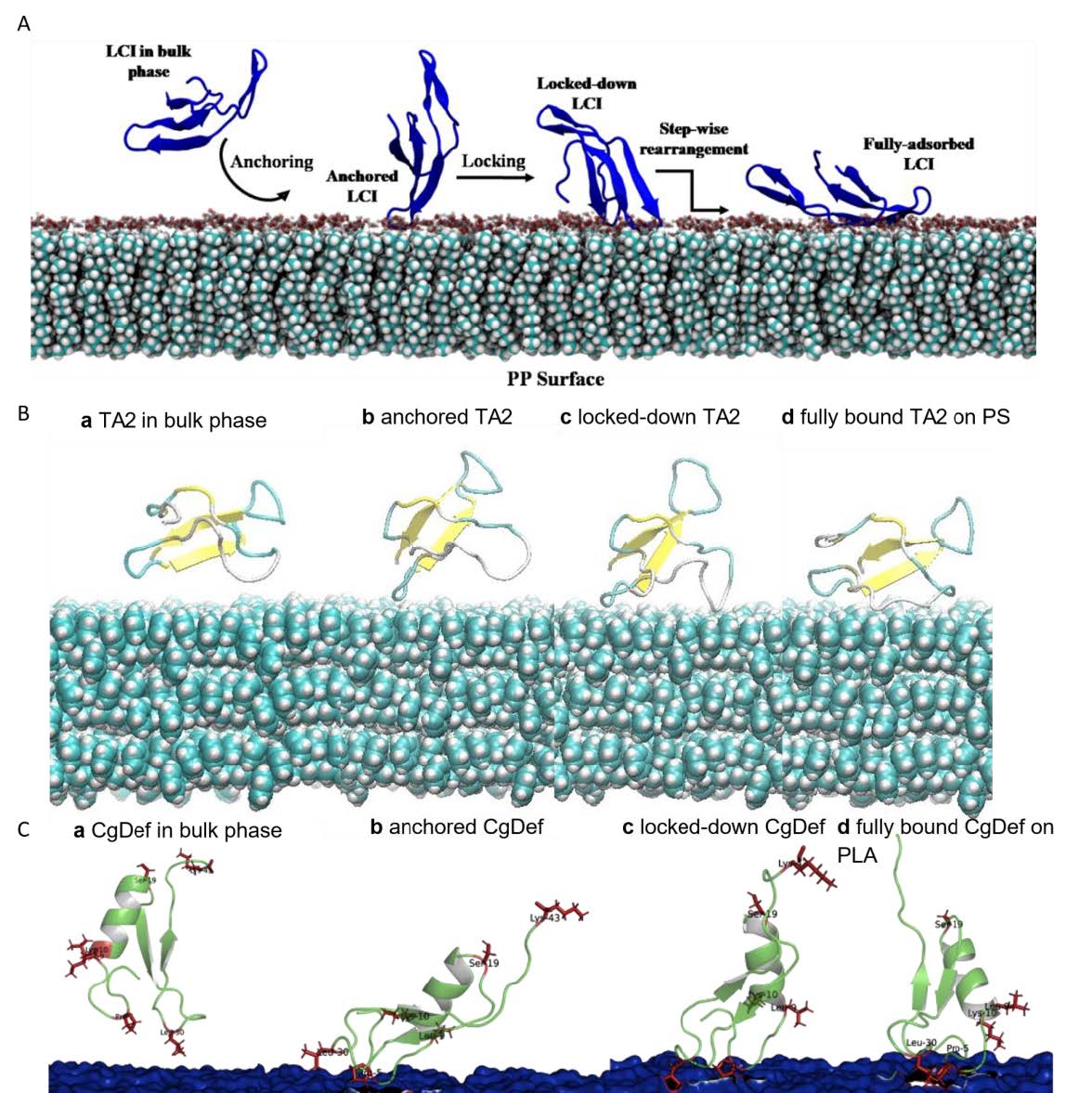 Figure 2: Elucidated binding process from anchoring, locking-down and full adsorption for APs on polymeric surfaces. A: LCI WT on PP surface. B: TA2 on PS surface. C: CgDef on PLA surface.
Figure 2: Elucidated binding process from anchoring, locking-down and full adsorption for APs on polymeric surfaces. A: LCI WT on PP surface. B: TA2 on PS surface. C: CgDef on PLA surface. Peptide adhesion promoters (anchor peptides, APs), due to their versatile binding capabilities to natural and synthetic polymers, are of high interest for many applications, including the design of novel plant health/protection agents, functionalization of a broad range of chemically different materials and surfaces, and targeted immobilization of enzymes for degradation of microplastic particles. APs can bind compound containers or antimicrobial peptides/enzymes specifically to natural surfaces such as cucumber, apple, barely, corn, and soybean leaves, synthetic polymers such as polystyrene (PS), polypropylene (PP), polyethylene terephthalate (PET), polyester-polyurethane (PUR), polycaprolactone (PCL), polylactic acid (PLA), metals such as stainless steel and gold and amorphous materials such as silicon wafers. To enable the rational design of tailor-made adhesion promoters for application requirements, a key aspect is a molecular understanding of the modes of interactions between peptides and material surfaces and its binding mechanism.The main goal of the PepSurSim project is to shine a light on the molecular interactions between APs and various synthetic and natural polymers. In particular, we aim to answer the following scientific questions at a molecular level: (i) How do APs bind to polymer surfaces? (ii) What is the binding mechanism of APs to polymers and particularly which predominant (driving) forces/interactions govern APs binding to polymers? (iii) How do charged amino acid substitutions influence the binding of APs to polymers? (iv) Can we generalize the learning to provide rational strategies to design peptides for improved surface binding or specific binding? The simulations of this project are tightly coupled to experiments done in our group to validate the proposed hypotheses and refine iteratively the computational models and parameters.
To this end, during the first CLAIX allocation period, we used all-atom MD simulations to understand the interactions between different APs (i.e., LCI, TA2, CgDef, and Macaque Histatin (MacHis)) and chemically diverse polymer surfaces (i.e., PP, PS, polylactic acid (PLA), PET, PUR, and cellulose). These preliminary results reveal the ability of MD simulations to explain the molecular basis for the improved binding of LCI charged substitutions to the PP surface. In the second phase of this project, we plan to extend our analysis to design in silico variants. The simulations planned for the extension of project, will provide the rationale required for future research planned on the design of APs with tailored anchoring properties (binding strength, specificity) to meet the application requirements.
Project Details
Project term
December 1, 2021–November 30, 2021
Affiliations
RWTH Aachen University
Institute
Jülich Supercomputing Centre
Project Manager
Principal Investigator
Methods
All MD simulations were performed using GROMACS2021.4 as available on the RWTH HPC clusters. Simulations were typically run for 50 ns, 100 ns or 300 ns on 8 nodes (96 cores in total). The new proposal we submit together with this report will request ~3.2 Million core-h.
Results
In the PepSurSim project, we modeled the following anchor peptides (AP)-polymer interactions using atomistic molecular dynamics (MD) simulations in three sub-projects:
- Liquid chromatography peak I of Bacillus subtilis (LCI) and polypropylene (PP)
- LCI and poly-lactic acid (PLA)
- CgDef and PLA
- Tachystatin A2 (TA2) and Cellulose
- TA2 and polystyrene (PS)
- Macaque Histatin (MacHis) and PP
Discussion
Sub-project 1:
The main goal of this sub-project was to develop and validate computational models and force field parameters for MD simulation of selected APs and polymer models. Different AMBER force fields were tested for their compatibility and the preservation of AP secondary structure during MD simulations. AMBER99SB-ILDN was generally best to maintain and reproduce the secondary structure of APs. GAFF parameters with RESP charges showed to reliably predict the structure and reproduce the experimental density of the polymers.
Sub-project 2:
The goal of this sub-project was to model binding mechanism and identify binding geometry of APs on polymers. MD simulations of LCI on PP, MacHis on PP, LCI on PLA, CgDef on PLA, and TA2 on cellulose, and PS were performed. We have analyzed the MD trajectories and proposed a binding mechanism and elucidated the role of amino acid substitutions in the binding of APs to polymers. Error! Reference source not found. shows the most common binding poses that were found for the interaction between various APs (LCI, CgDef, TA2 and MacHis) and natural (cellulose) and synthetic (PLA, PP) polymeric surfaces. Based on the analysis of the most common binding pose of each AP, we identified the most prominent binding residues within the AP. These binding residues are often important for binding and should not be altered in peptide design. In the new extension proposal, we apply for more computing time to finalize the missing deliverables from our initial plan. These include the simulation of the following AP-polymer pairs: LCI on PS and cellulose, TA2 on PP and PLA, MacHis on PS, PP and PLA and CgDef on PP, PS and cellulose. The final scope of AP-polymer pairs may vary based upon the experimental outcomes to judge the APs in vitro ability to bind to the suggested target surface. Through further study, we are able to distinguish binding patterns that occur per AP or per polymer- meaning that we aim to elucidate specific binding motifs or structural determinants that govern binding of APs to one polymer and possibly hinder or weaken binding to another. Drawing these conclusions requires atomistic knowledge about the binding process of each AP on the respective polymeric surface.We investigated the complete binding process of APs from fully solvated state until final adsorption onto the polymeric surface as shown in Figure 2. The following AP-polymer pairs were not delivered within the current project: LCI-PS, LCI-cellulose, MacHis-PS, MacHis-PLA, MacHis-Cellulose, TA2-PP and TA2-PLA. CgDef simulations were out of scope of the initial plan, but have been performed within this project due to great success on PLA.
With the extension proposal, we apply for more computing resources to answer the raised questions about the different spreading of APs on the polymer surface and to extend our AP-polymer pair collection. A larger library of simulated AP-polymer pairs allows us to draw conclusions more reliably and propose a generalized design strategy for peptide binding on a vaster variety of polymeric surfaces.
Sub-project 3:
The main objective of this sub-project is to predict and evaluate AP variants with tailored binding (in silico & wet lab). This goal was only partially achieved sofar. We developed a computational screening workflow to generate and evaluate the anchor peptides variants using the established screening system. In silico mutagenesis was carried out for LCI variants, CgDef variants. In silico mutagenesis is pending for further improved TA2 variants for PS and cellulose binding, a combined variant of CgDef as well as LCI variants on PLA and MacHis variants for improved pH-dependent PP binding. Furthermore, in silico mutagenesis shall be carried out for all not-yet delivered AP-polymer pairs as described in sub-project 2 based upon the outcome of the respective computational analysis.
We apply for more computational time to investigate the already proposed CgDef variants on the PLA surface, simulate and investigate the double variants and thereby refining our learnings from the LCI on PP investigation. This refinement is of importance as the surface characteristics of both studied polymers differ in their hydrophobicity. Where PP is purely hydrophobic and inert, PLA consist of a polar surface due to the presence of ester bonds. Investigation of not only hydrophobic but more and more hydrophilic surfaces, finally leading to the investigation of purely hydrophilic surfaces, will enable us to identify (i) key binding principles of AP to surfaces indifferent to the surface and (ii) which design strategies can be proposed based upon the hydrophobicity/hydrophilicity of the desired surface. These learnings may also enable us to ultimately design and engineer APs that can specifically bind and differentiate between different polymers.
Additional Project Information
DFG classification: 302-03 Chemical Solid State and Surface Research, Theory and Modelling
Cluster: CLAIX
Publications
We submitted the following paper for publication:
- A. Illig, N. Siedhoff, U. Schwaneberg, M.D. Davari, A hybrid model combining evolutionary probability and machine learning (MERGE) leverages data-driven protein engineering, doi:
https://doi.org/10.1101/2022.06.07.495081.
We are currently preparing following manuscripts for the publications: - K.-W. Hintzen, J. Mangler, L. Apitius, L. F. Stütgens, F. Jakob, M. D. Davari, U. Schwaneberg, Adsorption of Tachystatin A on a cellulose surface: A MD simulation study (To be submitted)
- K.-W. Hintzen, G. V. Dhoke, M. Sobota, F. Jakob, M. D. Davari, U. Schwaneberg, MD simulation reveals the binding mechanism and role of charged residues for binding of LCI on an inert polypropylene surface (To be submitted)
- S. Meng, H. Jung, M. D. Davari, U. Schwaneberg, Molecular understanding of an antimicrobial peptide (AMP) with PS polymer surface (To be submitted)