Project
Interatomic Potentials for Amorphous Solids From Second Principles
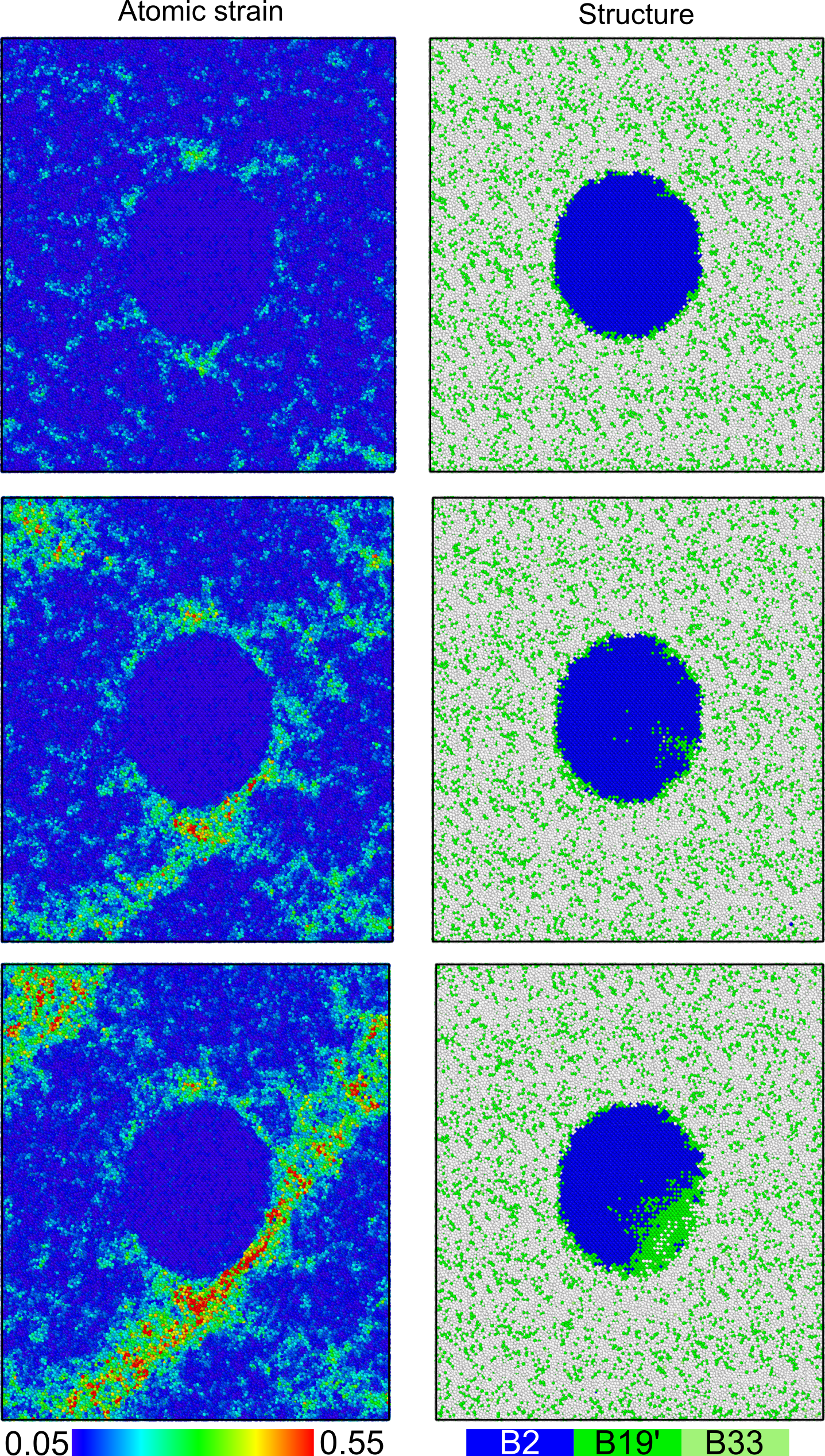 Figure 1: Shear band formation and structure evolution of a Cu50Zr50 matrix with a crystalline CuZr-B2 inclusion. The strain localizes at the interface until a shear band is formed. A martensitic phase transition from the B2 to the B19' structure of roughly the same width as the shear band can be observed in the crystalline phase.
Figure 1: Shear band formation and structure evolution of a Cu50Zr50 matrix with a crystalline CuZr-B2 inclusion. The strain localizes at the interface until a shear band is formed. A martensitic phase transition from the B2 to the B19' structure of roughly the same width as the shear band can be observed in the crystalline phase. 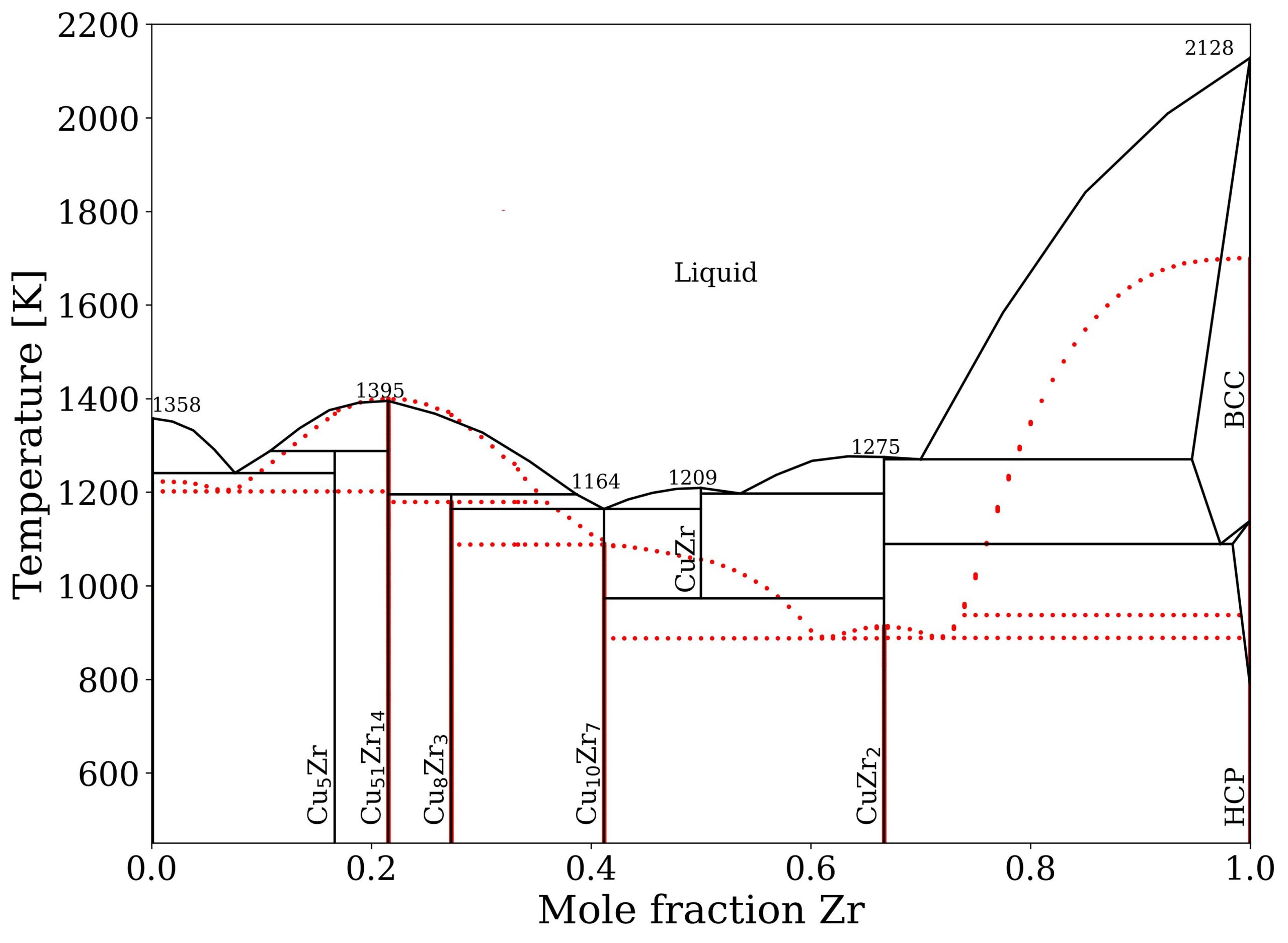 Figure 2: Phase diagram as determined by He et al. (black) [1] and calculated with the ACE potential (red).
Figure 2: Phase diagram as determined by He et al. (black) [1] and calculated with the ACE potential (red). 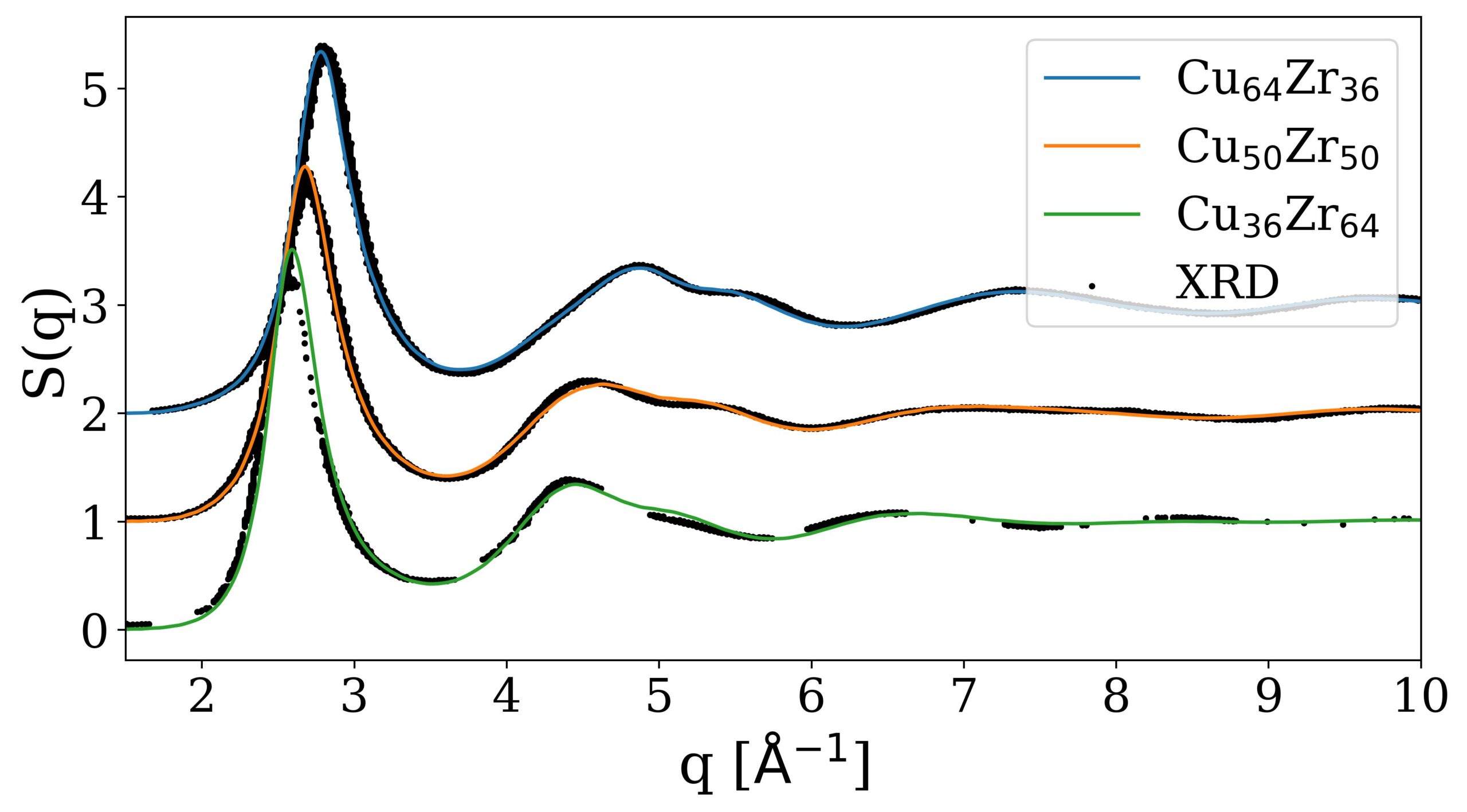 Figure 3: Total structure factor for simulated MG samples of varying compositions in comparison to X-ray diffraction data from Mattern et al. [2] .
Figure 3: Total structure factor for simulated MG samples of varying compositions in comparison to X-ray diffraction data from Mattern et al. [2] . Metallic glasses (MGs) can be produced by quickly quenching alloy melts. They typically show a higher elastic limit and strength than crystalline structures of the same material, but are very brittle and tend to fail catastrophically. Their ductility can be improved by including crystalline precipitates in a glassy matrix.
Atomistic simulations can help to understand the underlying structure-property relations. First-principles methods, however, are unfeasible because of their size and time limitations. Therefore, molecular dynamics (MD) simulations that use empirical inter-atomic potentials are the method of choice. Consequently, the accuracy of the simulations relies on the quality of these potentials. Here recent developments in the field of machine learning interatomic potentials allow massive improvements compared to classical potentials. They are fitted to energies and forces from density-functional theory (DFT) calculations. This allows to transfer the accuracy of first-principles to much larger scales. In this work we were interested in the Cu-Zr materials system, which forms a metallic glass over a wide compositional range. We fitted an atomic cluster expansion potential to a large set of training data produced with DFT and applied it in large scale molecular dynamics simulations.
Project Details
Project term
December 1, 2021–November 30, 2022
Affiliations
TU Darmstadt
Institute
Materials Modeling Division
Project Manager
Principal Investigator
Methods
DFT allows calculating energies and forces of an atomic structure at 0 K. It scales as N3 where N is the number of atoms in a system and is therefore limited to simulations of a few hundred atoms even on modern HPC systems. We used it to generate Cu-Zr training data and fitted an atomic cluster expansion potential to this data. Then the potential was applied in molecular dynamics simulations that integrate the Newtonian equations of motion to evolve a system in time. Their scaling is order N, and they allow simulating systems that contain millions of atoms.
Results
We tested our new potential by comparing it to previous potentials, DFT data and experiments. We found that it can reproduce the formation energies of intermetallic Cu-Zr phases with a much higher accuracy than previous potentials. Using the calphy code, which employs thermodynamic integration to obtain Gibbs free energies, we calculated a concentration-temperature phase diagram for our potential that agrees well with the experimental one. Amorphous structures produced by quenching Cu-Zr melts of various compositions showed that we can reproduce the experimental glass structure with high accuracy. For this we compared the total structure factors of our simulated structures with those measured using X-ray diffraction. To investigate them on an atomistic scale we used Voronoi analysis and found that the glass samples consist of a plethora of different Voronoi polyhedra. This contradicts results found with classical interatomic potentials, but agrees very well with available DFT and experimental data. Tensile tests of a Cu50 Zr50 glass with a B2-CuZr inclusion showed that the crystalline phase undergoes a martensitic phase transformation to the B19’ phase upon interacting with a shear band.
Discussion
Our results show that the ACE formalism allows a very accurate description of complex structures. The unmatched accuracy of formation energies allowed us to calculate a phase diagram that reproduces the experimentally determined one with a rather high accuracy. Shortcomings like the underestimated melting points clearly show the need for improved exchange-correlation functionals, as machine learning potentials rely on accurate training data. As shown for the glass structures the high accuracy allows determination of structural features on an atomistic scale. The tensile tests of glass-crystal compounds show that our potential can help to understand deformation mechanisms in MGs. In the future systematic large scale simulations could help to improve the ductility and corresponding applicability of MGs in structural applications.
Additional Project Information
DFG classification: 406 Materials Science
Software: LAMMPS, PACEMAKER, VASP
Cluster: Lichtenberg
Publications
Leimeroth, N. Reproducible, High-Throughput Potential Fitting in pyiron, Poster DPG Meeting Regensburg, Germany, 04.09-09.09 (2022)
Leimeroth, N. A general-purpose interatomic potential for the Cu-Zr system, The Minerals, Metals & Materials Society annual meeting / TMS, San Diego, USA, 19.03-23.03 (2023)
Citations
[1] He, X. C.; Wang, H.; Liu, H. S.; Jin, Z. P. Thermodynamic description of the Cu–Ag–Zr system, Calphad 30 (4) 367–374. (2006)
doi:10.1016/j.calphad.2006.09.001.
[2] Mattern, N.; Jóvári, P.; Kaban, I.; Gruner, S.; Elsner, A.; Kokotin, V.; Franz, H.; Beuneu, B.; Eckert, J. Short-range order of Cu–Zr metallic glasses, Journal of Alloys and Compounds 485 (1) 163–169. (2009)