Project
Insights on drug binding to HIV-1 TAR by well-tempered metadynamics
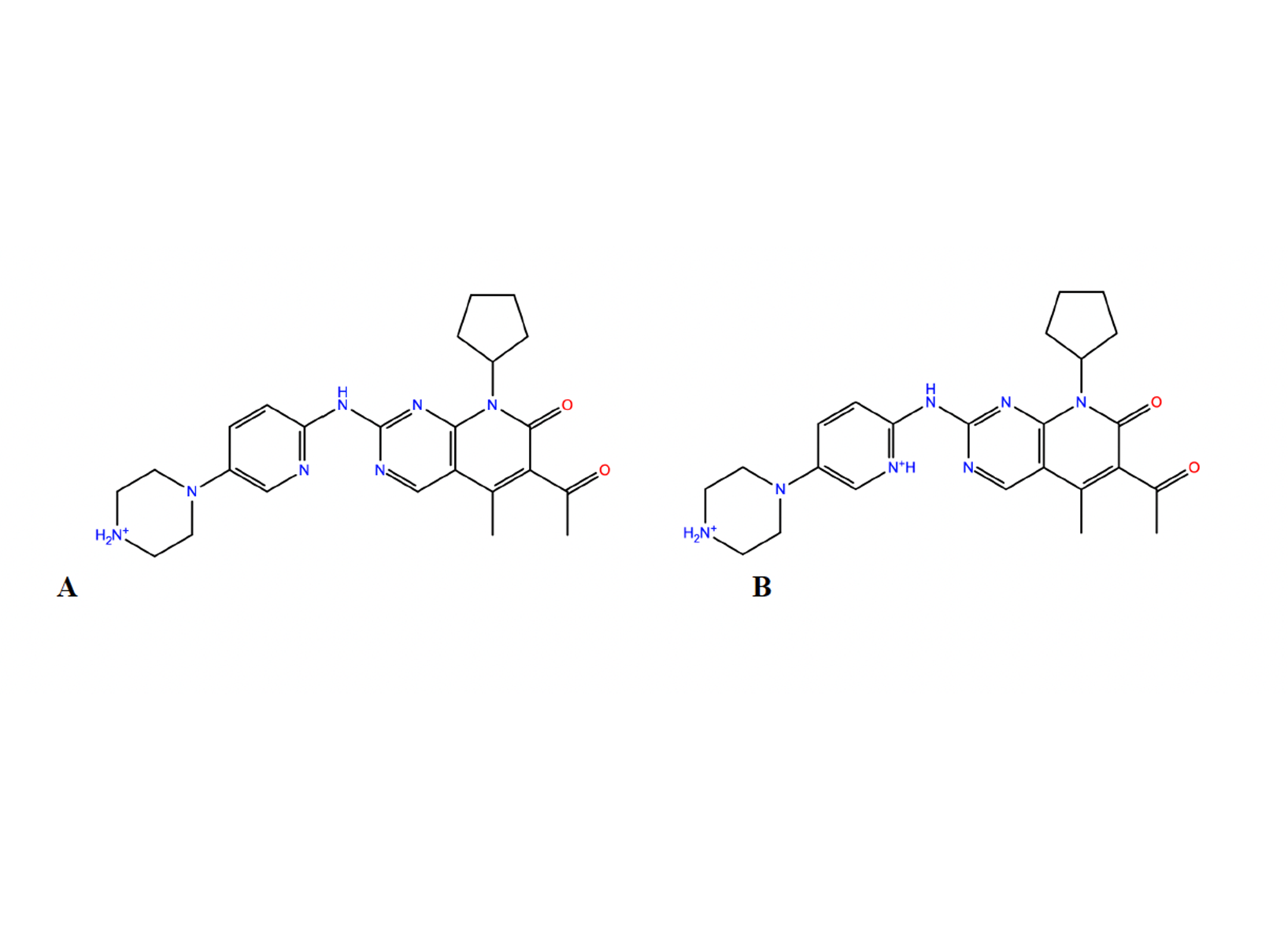 Figure 1. The two protonation states A (overall charge +1) and B (overall charge +2) of Palbociclib considered here.
Figure 1. The two protonation states A (overall charge +1) and B (overall charge +2) of Palbociclib considered here. 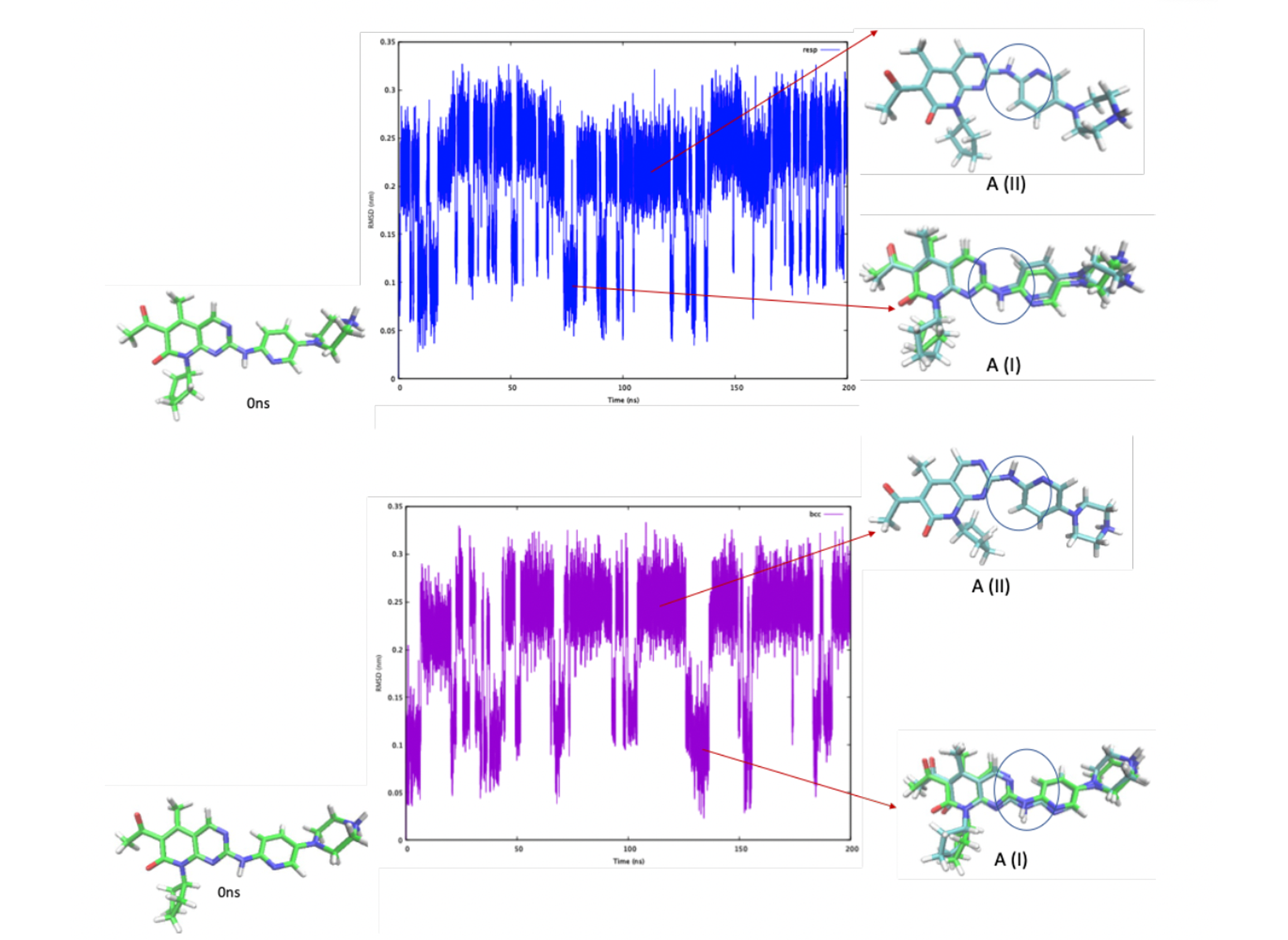 Figure 2. RMSD relative to the initial structure of A as a function of simulated time, with the RESP (top) and BCC
(bottom) parameterizations. The circle highlighted the NH linkage which has different directions in A-I and A-II.
Figure 2. RMSD relative to the initial structure of A as a function of simulated time, with the RESP (top) and BCC
(bottom) parameterizations. The circle highlighted the NH linkage which has different directions in A-I and A-II. 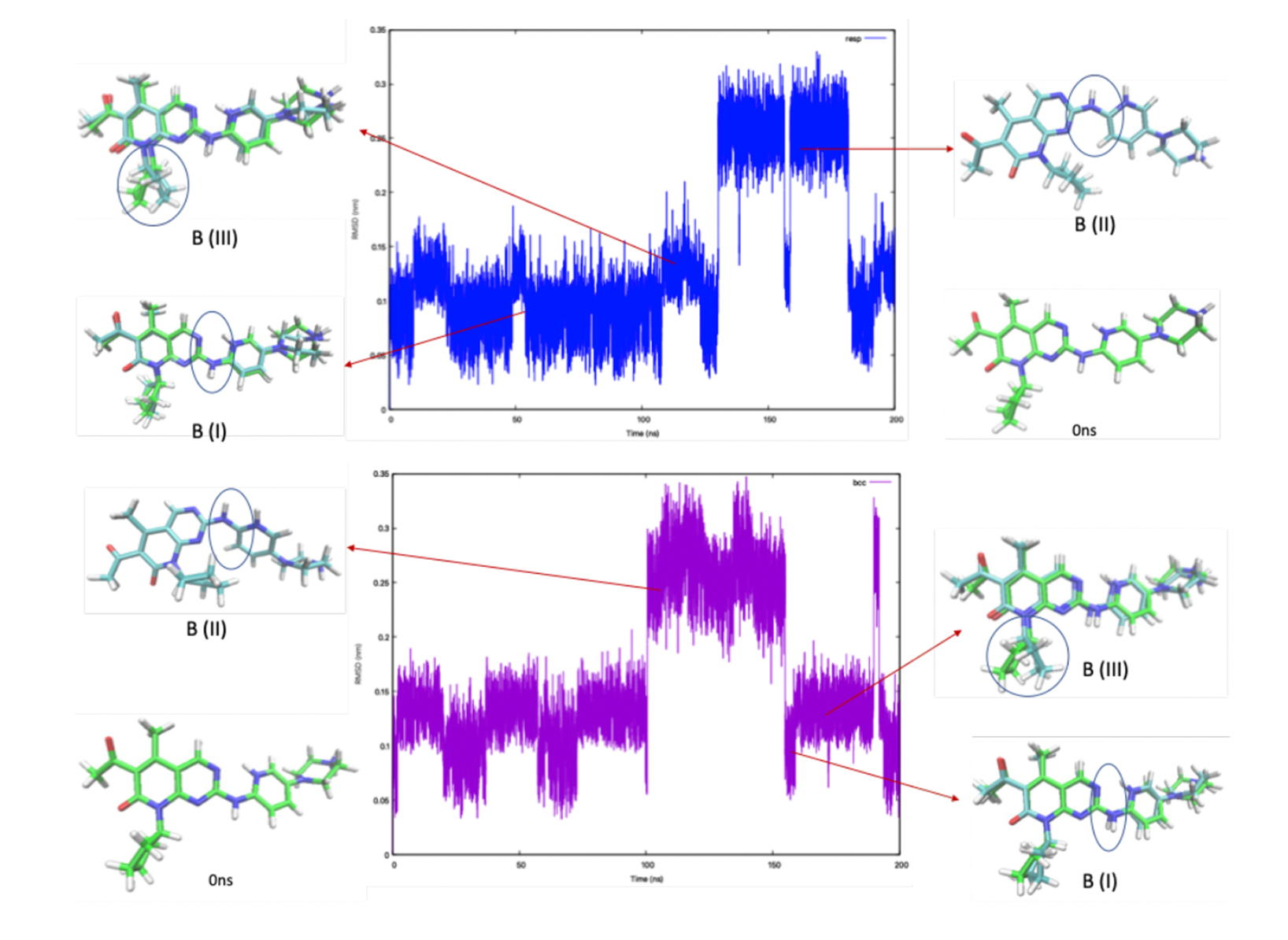 Figure 3. RMSD relative to the initial structure of B as a function of simulated time, with the RESP (top) and BCC
(bottom) parameterizations. The circle highlighted the NH linkage which has different directions in B-I and B-II, and
cyclopentane group in B-III.
Figure 3. RMSD relative to the initial structure of B as a function of simulated time, with the RESP (top) and BCC
(bottom) parameterizations. The circle highlighted the NH linkage which has different directions in B-I and B-II, and
cyclopentane group in B-III. 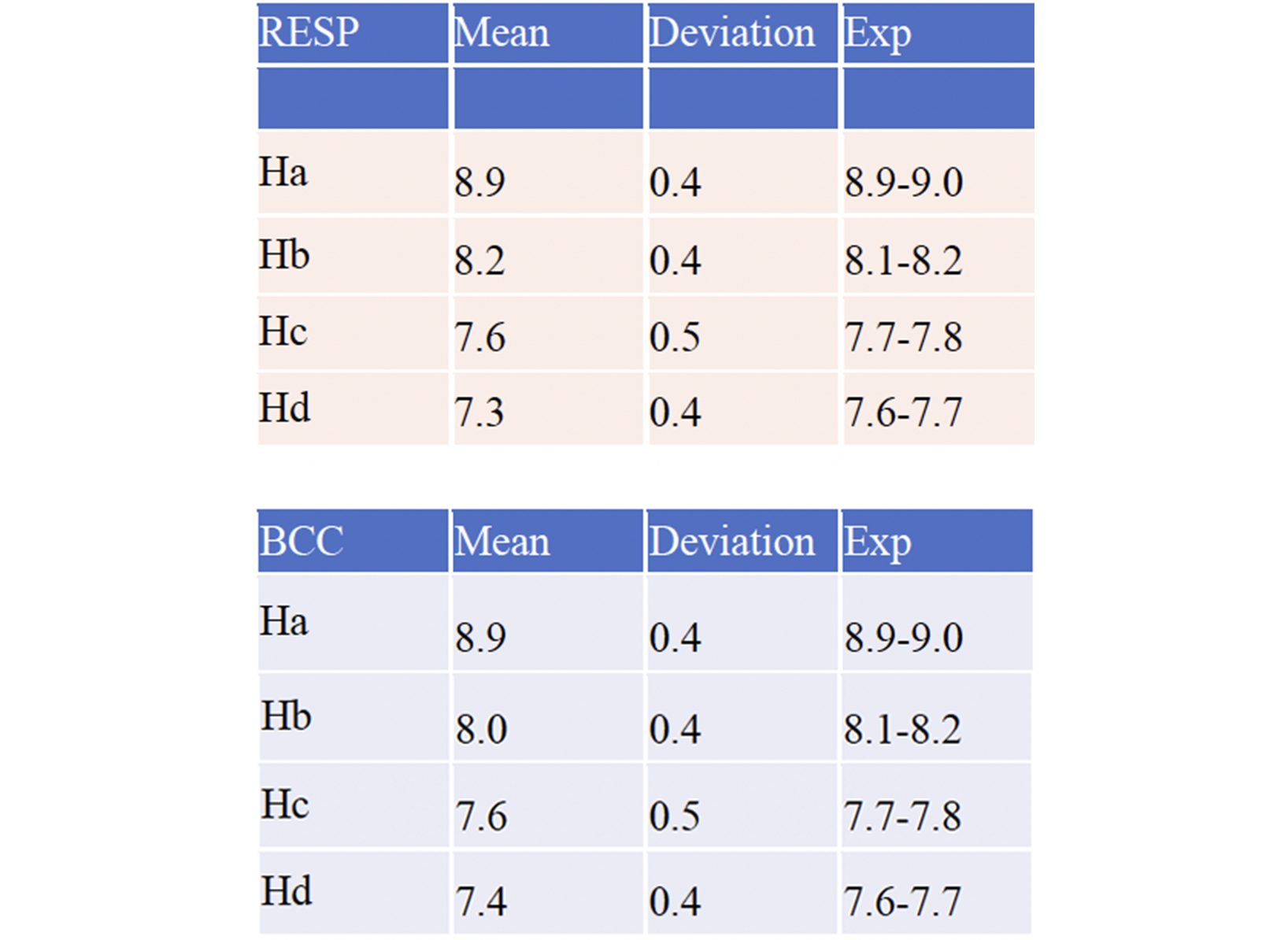 Table 1. Palbociclib’s chemical shifts from calculation (this work) and experiment at pH 7 (Matthew D. Shortridge,
Venkata Vidalala, Gabriele Varani, bioRxiv 2022)
Table 1. Palbociclib’s chemical shifts from calculation (this work) and experiment at pH 7 (Matthew D. Shortridge,
Venkata Vidalala, Gabriele Varani, bioRxiv 2022) 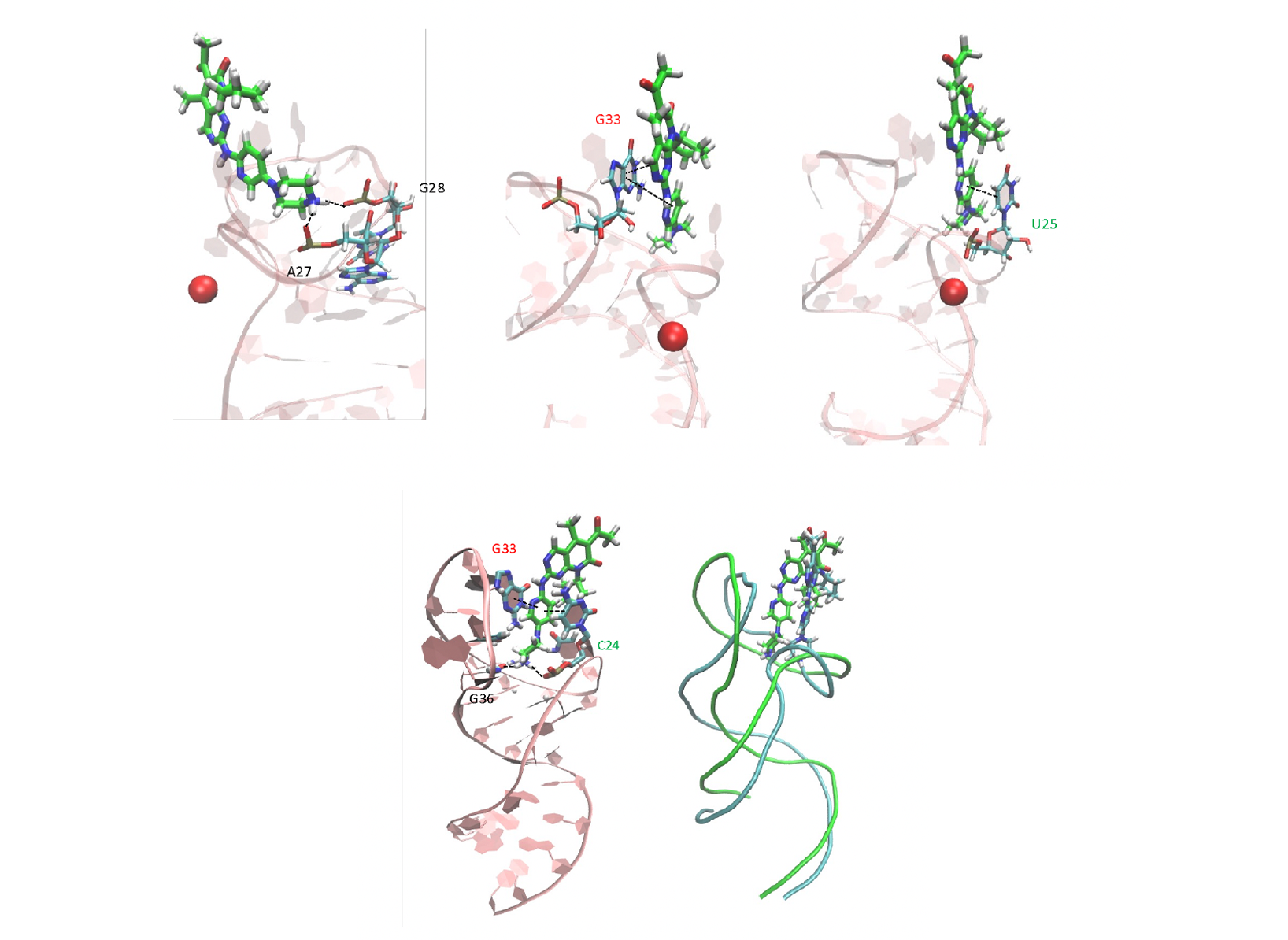 Figure 4. Palbociclib poses onto TAR as emerging from the MD with the Mg(II) ion (top) and without the ion (bottom,
left) . In the top view, three different orientations of the ligand binding to TAR are shown for the sake of clarity.
Bottom, right: Superposition of the two poses (RMSD= 4.2 Å).
Figure 4. Palbociclib poses onto TAR as emerging from the MD with the Mg(II) ion (top) and without the ion (bottom,
left) . In the top view, three different orientations of the ligand binding to TAR are shown for the sake of clarity.
Bottom, right: Superposition of the two poses (RMSD= 4.2 Å). 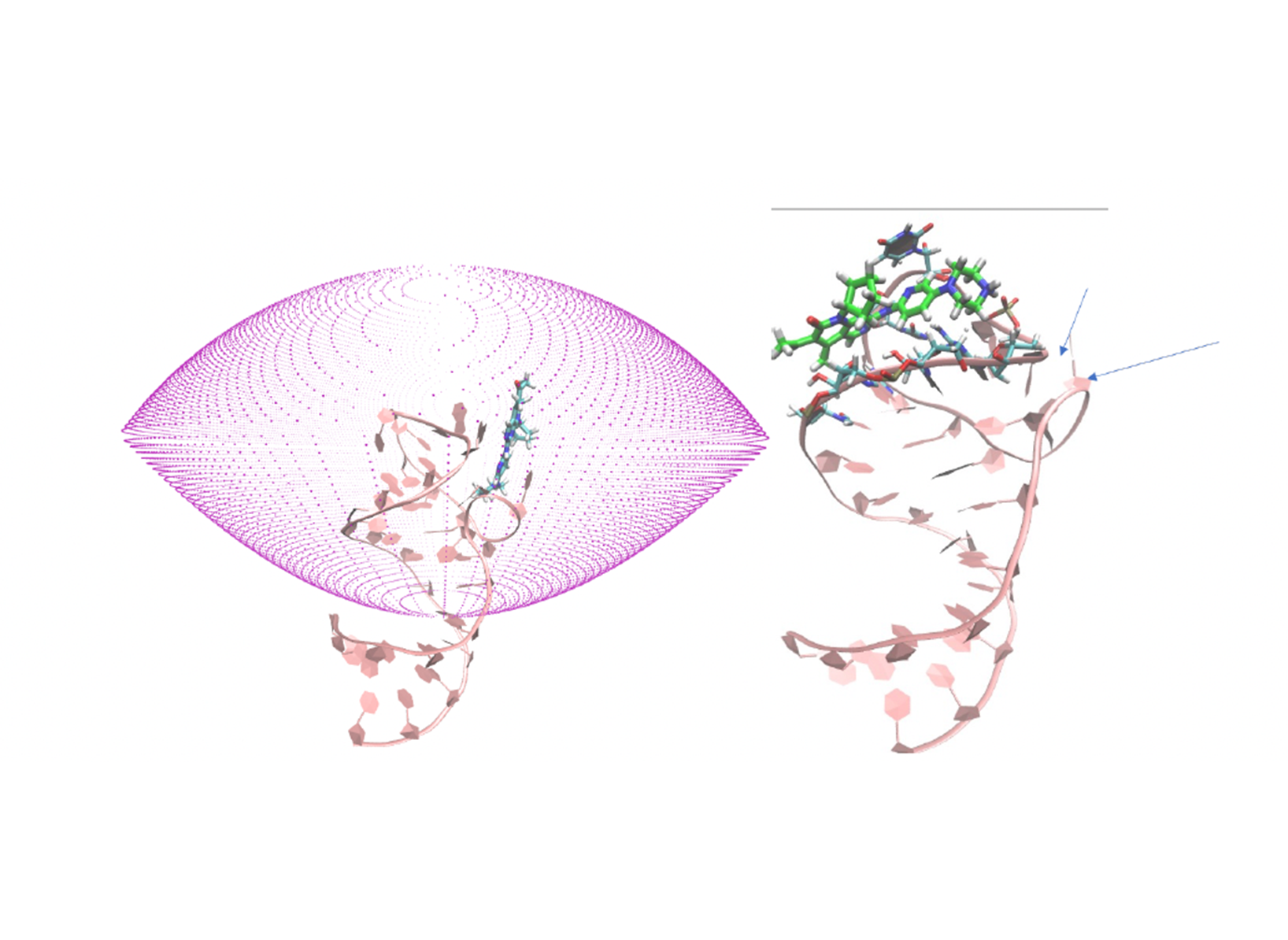 Figure 5. Left: Parabolic-solid volume for LV-MetaD; Right: A frame from the simulation showing the loop and the
bulge always close to each other (indicated by arrows), preventing ligand from going back to the initial pose.
Figure 5. Left: Parabolic-solid volume for LV-MetaD; Right: A frame from the simulation showing the loop and the
bulge always close to each other (indicated by arrows), preventing ligand from going back to the initial pose. The fundamental role of RNA for cell dysfunction associated with diseases has called upon pharmacologists to develop small ligands targeting RNA instead of proteins. Unfortunately, however, even FDA-approved drugs feature mediocre affinities for their targets, and, thus, new therapeutic agents binding stronger to RNA than in the past are strongly needed. Recently, our experimental collaborator Prof. Gabriele Varani (University of Washington at Seattle, US) has shown that a blockbuster drug – the kinase inhibitor Palbociclib – binds the HIV-1 transactivation responsive (TAR), a very important target for antiviral infection, in the 100 nM range. Unfortunately, the structural determinants of the RNA/drug complex are not known. Predicting poses and free energy of binding of palbociclib to TAR could help design derivatives with high affinity towards this target.
Our team has used molecular dynamics simulations, along with enhanced sampling approaches such as metadynamics, to investigate structure, dynamics of TAR (Musiani et al, JACS, 2014) as well as ligand binding to it (Do et al., JCTC, 2012, Do et al., JCTC, 2013), in collaboration with Prof. Varani. Here we have built on our expertise to get insights on structure, spectroscopic properties and energetics of palbociclib in water and bound to TAR. Comparison was made with NMR experiments carried out in Varani’s lab.
Project Details
Project term
February 1, 2023–January 1, 2024
Institute
Computational Biomedicine Institute
Principal Investigator
Methods
Palbociclib in water
System. The structure of the ligand was taken from PubChem library (https://pubchem.ncbi.nlm.nih.gov/compound/Palbociclib). Hydrogen atoms were added assuming standard bond lengths and bond angles. Two groups are tritable in the molecule: the piperazine nitrogen and the pyridine nitrogen atoms. The pKa of the piperazine nitrogen in palbociclib is 7.4 (PubChem), so the nitrogen is expected to be protonated at physiological pH. The pKa of the pyridine nitrogen is 3.9 (PubChem), so it is expected to be deprotonated (Fig. 1A) at physiological pH and protonated in acid conditions (Fig. 1B). Here we consider both protomers. They were solvated in a periodic octahedron box with approximately 2,300 water molecules. 10 sodium cations, 3 potassium cations and 14 chloride anions were added to neutralize the system and mimic the experimental ionic concentration with 200mM NaCl and 50mM KCl (Matthew D. Shortridge, Venkata Vidalala, Gabriele Varani, bioRxiv 2022). Molecular dynamics. The partial charges of A and B were calculated using semi-empirical BCC (Jakalian et al., J. Comput. Chem, 2002) or ab initio RESP (Wang et al., J. Comput. Chem., 2000) schemes. The other parameters of the ligand were generated with General AMBER Force Field – GAFF (Wang et al., J. Comput. Chem, 2004). The water and ions’ force fields were the TIP3P (Jorgensen et al., J. Chem. Phys., 1983) and (Joung and Cheatham, JPC B, 2008), respectively. PME (Essmann et al., J. Chem. Phys., 1995) was used for long-range electrostatics interactions. van der Waals interactions and the real part of electrostatic interactions were truncated at 12 Å. All MD simulations of the two protomers in water were performed in 200 ns at temperature 310 K and constant pressure of 1 bar (same as that of Varani’s experiments, Matthew D. Shortridge, Venkata Vidalala, Gabriele Varani, bioRxiv 2022). Constant temperature and pressure conditions were achieved by the Nose-Hoover thermostat (Evans and Holian, J. Chem. Phys., 1985) and the isotropic Parrinello−Rahman barostat (Parrinello and Rahman, J. Appl. Phys., 1981), respectively. The timestep was set to 2fs. NMR chemical shift calculations were performed using a QM/MM scheme based on the ORCA code (F. Neese et al., J. Chem. Phys., 2020) on 200 MD snapshots. The results were compared with the NMR data from our collaborator (Matthew D. Shortridge, Venkata Vidalala, Gabriele Varani, bioRxiv 2022).
TAR-Palbociclib complex
According to the experimental conditions, one Mg(II) ion may or may not be present and interact with the bulge of RNA (Varani, personal communication): the binding affinities are not very different between the two cases. We considered both here. The structure of the complexes was prepared by a docking procedure using the program Glide from Maestro, Schrödinger (Yang Y et al., J. Chem. Theory Comput. 2021, Friesner R. A et al., J. Med. Chem. 2006, Halgren T. A et al., J. Med. Chem. 2004). The protomer A was used. Two force field parameter sets to describe TAR and ions, one is AMBER14sb + bsc1 force field (Ivani et al. Nat. Methods 2015, Maier et al. J Chem Theory Comput. 2015), the other is the new force field parameters developed and revised by the D. E. Shaw Research group (New York, United States) called DES-Amber (Tucker M R. et al., J. Phys. Chem. B 2022). Along with AMBER14sb + bsc1 force field, the same force field parameters as above were used for the water and the ions. In case of DES-Amber force files, the ions were parameterized in different ways, and the water model TIP4P-D was
employed. For the ligand, we selected the RESP parametrization. We performed four 1 microsecond-long MD simulations, in the same conditions as the simulations above, on the following systems:
1. Adduct with Mg(II) (AMBER force field
2. Adduct without Mg(II) (AMBER force field)
3. Adduct with Mg(II) (DES-Amber force field)
4. Adduct without Mg(II) (DES-Amber force field)
MD simulations on the 3. and 4. lead to partial or complete dissociation of the ligand (not shown) and were discarded. One of the final snapshots of the MD of 1 and 2 were used as the starting structures for the Well-tempered Metadynamics simulations (WTMetaD) (Barducci et al., Phys Rev Lett 2008 and Dama et al., Phys Rev Lett 2014). This provides the free energy surface of molecular systems as a function of wise-chosen degrees of freedom, called collective variables (CVs). Here, WTMetaD was employed to study the binding of ligand Palbociclib to TAR. One of the challenges here is the choice of appropriate collective variables (CVs) that can describe the slow motions of the system. Different setups were used, but they all failed (see results).
Results
Palbociclib in water
The protomers A and B showed the interchange between different conformations. A exhibits two major conformations irrespectively of the RESP/BCC parametrization, corresponding to two separated parts in the RMSD graph (Fig 2). In one conformation (A-I) , the NH linkage points in the same direction as the ring’s CO group1, while in the other (A-II, RMSD= 2.3 Å), it points in the opposite direction. The 1H chemical shifts calculated for A compare well with experiments (Table 1). The simulations using BCC and RESP give almost the same results. B exhibits two conformations similar to those of A, and, in addition, one that differs from A-I by the conformation of the cyclopentane group (B-III, Fig.3).
Palbociclib/TAR complex
During the dynamics (using the AMBER force field, see Methods), the ligand (in its conformation A-II, see Fig. 2-3) was accommodated by TAR forming hydrogen bonds and pi stacking interactions in a similar fashion either with Mg(II) (Fig. 4, top) or without the ion (Fig. 4, bottom, left). It contacted some nucleotides in the loop and the bulge and nearby nucleotides of the RNA via hydrogen bonds and pi stacking. One of the final snapshots of the MD was used as the starting structure for the WTMetaD simulations. First, we used a variant of WTMetaD called Localized Volume-based Metadynamics (LV-MetaD). In this approach, the motions of the ligand are restricted inside a parabolic-solid volume including the important parts for binding and neighboring regions, and we employ three system-independent CVs based on the volume parameters to deposit the bias potential. ~800 ns of trajectory were collected in this simulation, however, we could not reach the convergence of the free energy profile of binding.
Inspecting the trajectory, we conclude that the ligand does not come back to the initial binding pose because the loop and the bulge are closing during the simulation. Fig. 5 shows the example for the System with the Mg(II) ion.
We then tested different CVs:
- (1 CV) Coordination number: between ligand and RNA; between ligand and nucleotides of
the bulge and the loop - (2 CV) Distance between ligand-RNA – Number of hydrogen bonds
- (2 CV) Distance between ligand-RNA – RMSD of bulge and loop
- (2 CV) Distance between ligand-RNA – Distance between bulge and loop
- (2 CV) Distance between loop and bulge – Coordination number
Unfortunately, none of those CVs worked for the ligand-RNA system, whether Mg(II) was present or not. No convergence could be obtained at the end for all WTMetaD simulations we tried. In most cases, the ligand came back and forth to interact with RNA but it could not find a stable pose, and the RNA was deformed during its interactions with the ligand.
Discussion
We have presented MD and metadynamics simulations of the Palbociclib drug in water and in complex with TAR. In the first set of simulations, the ligand turns out to experience different conformations during the dynamics. Our calculated NMR chemical shifts at pH 7 are comparable with experiment, validating our simulations. Our calculations also predict that acidification does not alter significantly the conformational space of the ligand, as the conformation explored are either basically the same observed at pH 7 or exhibit small changes. Our well-tempered metadynamics did not provide insights on the binding of the ligand to TAR. Among the possible reasons that may explain our unsuccessful attempts, we mention here some limitations of the force field used to describe TAR/ligand interactions. more accurate approaches, such as QM/MM simulations (in which the drug is treated at the QM level and the RNA and the solvent at the classical level) might be required to investigate this system. Such simulations are planned in our lab. Alternatively, experimental data (such as NMR data) could be integrated into the simulations to guide the simulations and improve the results.
Additional Project Information
DFG classification: 201-02 Biophysics
Software: Gromacs, ORCA, PLUMED
Cluster: CLAIX
Publications
This work will appear in the PhD thesis of PhD student Gia Linh Hoang.