Project
High-Throughput Design of Chalcogenide Materials for Optoelectronic Devices
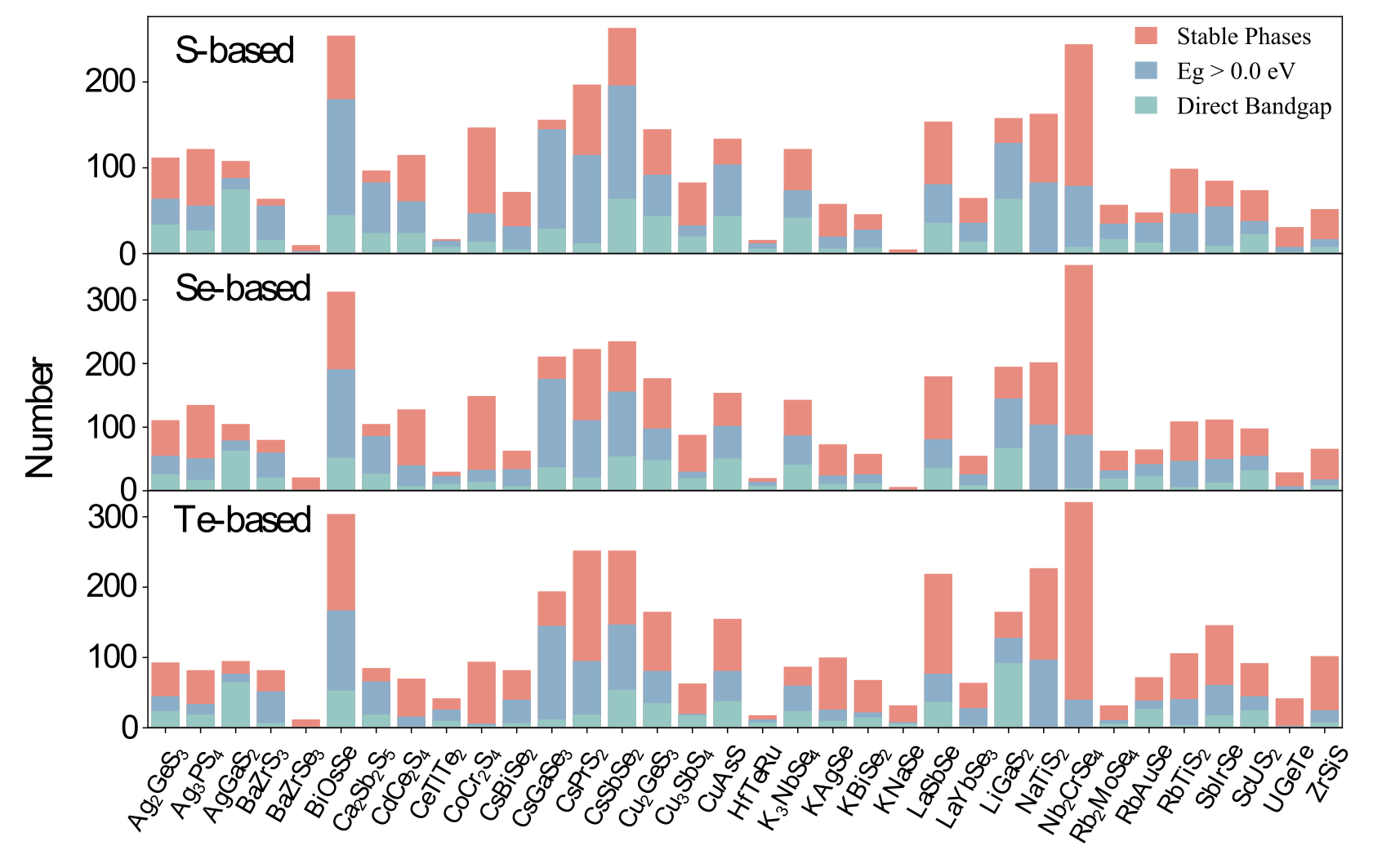 Figure 1: Statistics on different properties of S-, Se-, and Te-based candidates in terms of structural prototypes. Red, blue, and green colors denote the number of stable phases, nonmetallic phases, and direct band gap phases, respectively.
Figure 1: Statistics on different properties of S-, Se-, and Te-based candidates in terms of structural prototypes. Red, blue, and green colors denote the number of stable phases, nonmetallic phases, and direct band gap phases, respectively. Optoelectronic materials have attracted significant attention owing to the global energy shortage and environmental concerns suffered from the excessive consumption of fossil fuels. By absorbing solar energy, which acts as a clean and sustainable energy source, the internal electric current can be produced and delivered based on the photoelectric effect.
The past several decades have witnessed a phenomenal development of the discovery and the performance modification on a series of optoelectronic materials, e.g., silicon and CdTe, etc. Recently, it has been demonstrated that the processes of novel material discovery and the prediction of their properties can be significantly accelerated by using advanced quantum mechanical methods based on density functional theory, high-throughput (HTP) techniques, and material databases. Compared to the conventional methods using the experimental trial-and-error strategy, this computational screening process can exhibit higher efficiency and more clear structure-property relationships.
In this work, we chose 34 ternary chalcogenide prototypes as initial entries to explore novel crystalline semiconductors with potential applications in photovoltaics, a prominent field within the realm of optoelectronics. In combination with the strategy of cation substitution, we built a material database containing 400,000 candidates. After a series of screening processes oriented by photovoltaic properties, including thermodynamic stability, band gap, electronic structure, and efficiency, a total number of 52 candidates were identified as promising absorber materials for solar cell devices. Our theoretical study provides a guideline for further experimental research focusing on the synthesis and validation of novel optoelectronic materials.
Project Details
Project term
May 1, 2022–April 30, 2023
Affiliations
TU Darmstadt
Institute
Theory of Magnetic Materials
Project Manager
Principal Investigator
Methods
The first-principles calculations were performed using an in-house developed HTP calculation environment. All the calculations are carried out within the DFT framework, as implemented in the Vienna ab initio simulation package. The interaction between core and valence electrons was explained using the frozen-core projector augmented wave pseudopotentials. In our calculations, the generalized gradient approximation with Perdew-Burke-Ernzerhof functional was used as the exchange-correlation functional. The crystal structures were fully optimized with the energy convergence criteria of 1 × 10−5 eV. The kinetic energy cutoff for plane-wave basis was set to 500 eV, in conjunction with a grid spacing of 2π × 0.03 Å−1 for Brillouin zone sampling.
Results
We implemented a systematic high-throughput screening process combined with first-principles calculations on ternary chalcogenides using 34 crystal structure prototypes. We generated a computational material database containing over 400,000 compounds by exploiting the ion-substitution approach at different atomic sites with elements in the periodic table. The thermodynamic stabilities of the candidates were validated using the chalcogenides included in the Open Quantum Materials Database. Then, we identified 1022 stable candidates and 7990 metastable candidates. Moreover, we trained a model based on Crystal Graph Convolutional Neural Networks to predict the thermodynamic stability of novel materials. Furthermore, we theoretically evaluated the electronic structures of the stable candidates using accurate hybrid functionals. A series of in-depth characteristics, including the carrier effective masses, electronic configuration, and photovoltaic conversion efficiency, was also investigated.
Discussion
In summary, we have presented a comprehensive high-throughput screening process based on the idea of ion substitution and ab initio calculations to explore promising ternary chalcogenides for optoelectronic applications. By replacing A- and B-site cations in ternary Ax By Xz chalcogenides crystallized in 34 structural prototypes, a rich material database containing > 400,000 Ax By Xz chalcogenide candidates was constructed. After a series of systematic first-principles calculations, we found 7943 unknown materials exhibiting satisfied phase stability against chemical decomposition. On this basis, the chemical distribution of phase stability in terms of the structural prototype and chemical composition was established, respectively. Furthermore, an effective stacked ensemble model based on machine learning algorithms was generated to predict the phase stability directly. By using the developed self-consistent hybrid functional, a photovoltaic-functionality-oriented material screening process involving > 6000 stable and metastable candidates was performed, providing a final identification of 52 chalcogenides as promising absorbers in single-junction solar cells. These materials exhibited good thermodynamic stabilities (Ef < 0 eV and ∆Eh < 50 meV/atom), suitable band gaps (1.1-1.5 eV), direct or quasi-direct electronic band structures (△Eg < 10 meV), ultra-low carrier effective masses (< m0 ), and high efficiencies (> 32 %). In particular, a series of materials represented by Sn3 BaSe4 show similar optical characteristics as CH3 NH3 PbI3 owing to the intrinsic lone-pair ns2 -cation-containing electronic configuration, which has been verified as an origin for the high conversion efficiency of the solar cell devices fabricated by Pb-based halide perovskites. Our work provides a promising routine for exploring novel ternary chalcogenide semiconductors with superior performances for optoelectronic applications. And the subsequent experimental efforts aiming at the synthesis and performance characterization are strongly called for.
Additional Project Information
DFG classification: 406 Materials Science
Software: VASP, Sumo, BoltzTrap, AutoGluon-Tabular framework
Cluster: Lichtenberg
Publications
Shen, C., Li, T., Zhang, Y., Long, T., Fortunato, N.M., Liang, F., Dai, M., Shen, J., Wolverton, C. and Zhang, H. Accelerated Screening of Ternary Chalcogenides for 4 Potential Photovoltaic Application. J. Am. Chem. Soc., 145, 40, 21925–21936 https://doi.org/10.1021/jacs.3c06207 (2023)