Project
Plasticity of Complex Crystals by Density Functional Theory (DFT)
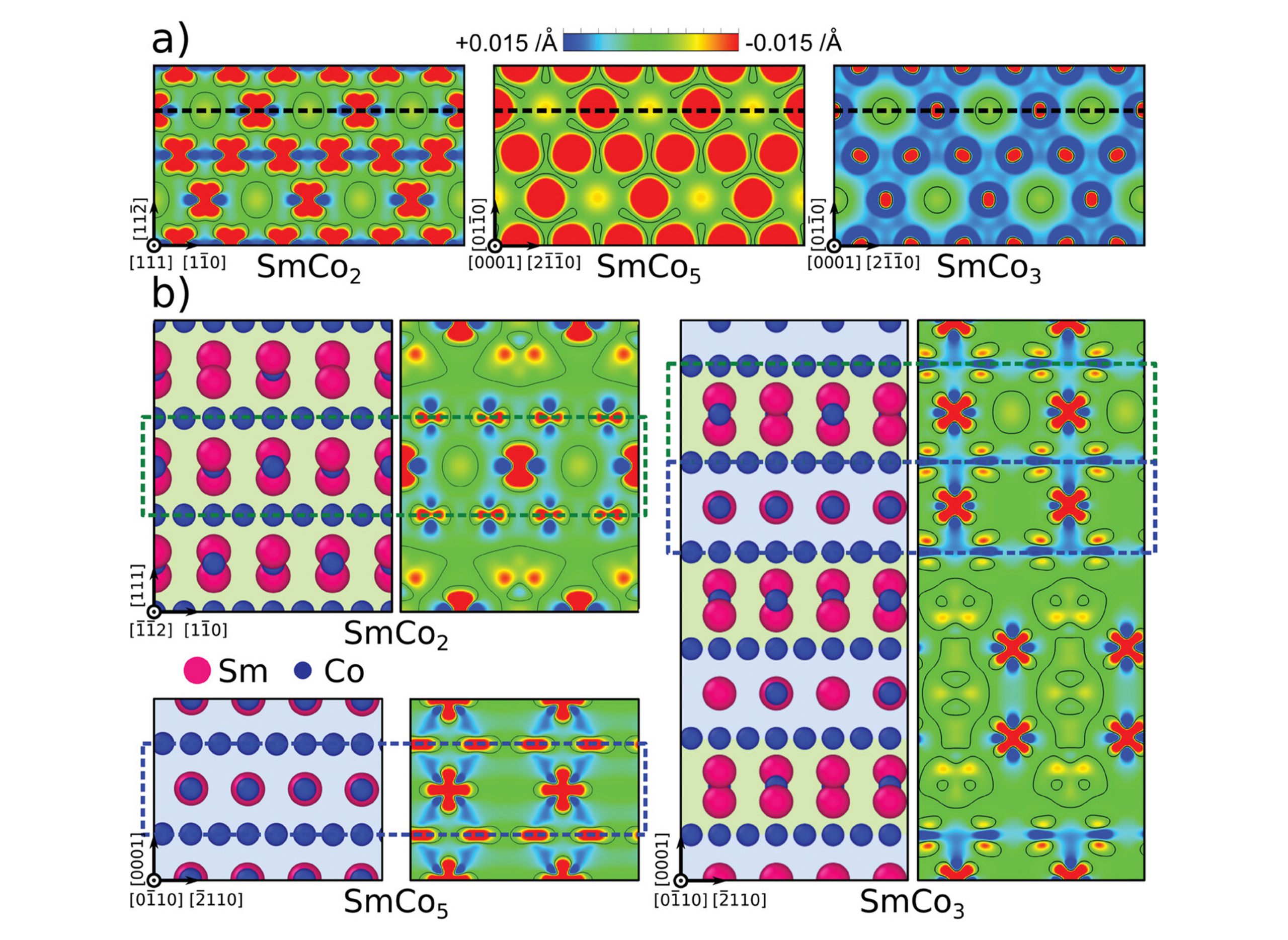 Charge density difference profiles of a) Co Kagomé-nets along {1 1 1} or basal planes and b) {1 1 2} or prismatic atomic layers (annotated as dotted lines in a)) in the SmCo2, SmCo5, and SmCo3 crystals. Regions of charge concentration and deficiency are displayed in red and blue respectively, and green areas represent a metallic or delocalized bonding environment. The fundamental building blocks in the SmCo3 crystal are indicated by different background colors and the equivalent {1 1 2} and prismatic atomic layers are marked using dashed boxes.
Charge density difference profiles of a) Co Kagomé-nets along {1 1 1} or basal planes and b) {1 1 2} or prismatic atomic layers (annotated as dotted lines in a)) in the SmCo2, SmCo5, and SmCo3 crystals. Regions of charge concentration and deficiency are displayed in red and blue respectively, and green areas represent a metallic or delocalized bonding environment. The fundamental building blocks in the SmCo3 crystal are indicated by different background colors and the equivalent {1 1 2} and prismatic atomic layers are marked using dashed boxes. 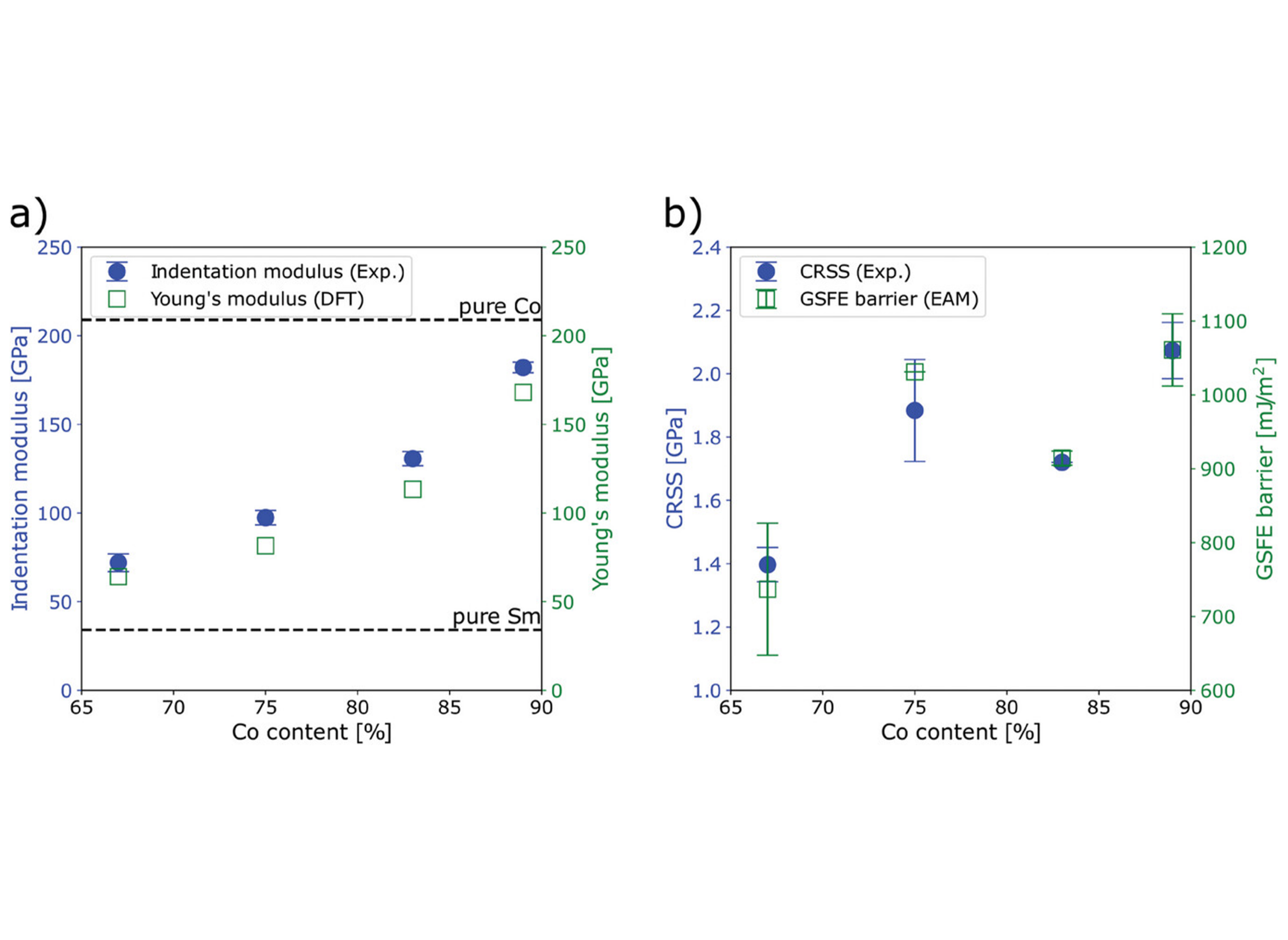 Comparisons of a) experimentally measured indentation moduli and DFT-calculated Young's moduli of Sm–Co intermetallics, and b) CRSS obtained from micropillar compression experiments and GSFE barriers calculated using the EAM potential.
Comparisons of a) experimentally measured indentation moduli and DFT-calculated Young's moduli of Sm–Co intermetallics, and b) CRSS obtained from micropillar compression experiments and GSFE barriers calculated using the EAM potential. In the proposed project, we will investigate the mechanisms at various active slip planes detected on select Sm-Co and Ta-Fe phases. First-principles simulations using Density Functional Theory (DFT) will be used to calculate the elastic property as well as produce an energetic comparison of various slip mechanisms in order to gain insight on the deformation mechanisms of complex intermetallics. Previous studies have been conducted to identify families of crystals which can model the “building blocks” and the “complex, larger unit cells” which contain structural semblance to a binary assembly of a pair of “building blocks”. The experimental counterpart of this project will conduct the nanoindentation and micropillar compression experiments, as well as visual characterization through high-resolution transmission electron microscopes (HR-TEM), to gain insight on all the slip systems occurring on the structures. These experiments will guide the direction of DFT simulations and computational analyses.
Project Details
Project term
February 2, 2024–April 10, 2025
Affiliations
RWTH Aachen University
Institute
Institute for Physical Metallurgy and Materials Physics
Principal Investigator
Methods
Density Functional Theory (DFT) implemented within the Vienna Software Package (VASP) code will be used for all electronic structure calculations, including geometry optimization, static, as well as transition state calculations. As such, there are a number of parameters that we need to benchmark before simulating each solid-state system. For each electronic structure calculation performed in DFT, the wavefunction of the system is determined iteratively through an SCF cycle. The SCF cycle is largely based on the Variational Principle that the ground-state energy of a structure is bounded by a minimum value. As such, calculations in DFT can be conducted without prior knowledge of the system’s wavefunction. Parallelization can be achieved using the message-passing techniques (MPI) implemented in VASP. For analysis and visualization we will mainly use OVITO and VESTA.
Results
Stollenwerk, T., Huckfeldt, P. C., Ulumuddin, N. Z. Z., Schneider, M., Xie, Z., \& Korte‐Kerzel, S. (2025). Beyond Fundamental Building Blocks: Plasticity in Structurally Complex Crystals. Advanced Materials, 37(6), 2414376.
This study examines the hypothesis that deformation behavior can be transferred from fundamental building blocks to structurally related phases using the binary samarium-cobalt system. SmCo2 and SmCo5 are investigated as fundamental building blocks and compared them to the structurally related SmCo3 and Sm2Co17 phases. Nanoindentation and micropillar compression tests are performed to characterize the primary slip systems, complemented by GSFE calculations via atomic-scale modeling (see Figure 1). The results show that while elastic properties of the structurally complex phases follow a rule of mixtures, their plastic deformation mechanisms are more intricate, influenced by the stacking and bonding nature within the crystal’s building blocks, as previously shown in Figure 1. These findings underscore the importance of local bonding environments in predicting the mechanical behavior of structurally related intermetallics, providing crucial insights for the development of high-performance intermetallic materials.
Schneider, M., Deformation mechanisms of Sm-Co intermetallics via ab-initio and atomistic modelling, Masterarbeit, RWTH Aachen, (04.07.2024 – 06.01.2025)
In this work, molecular statics MS and ab-initio DFT
simulations are employed to exhaustively investigate the active slip systems identified by Stollenwerk et al. through GSFE calculations and NEB methods. Atomic path visualizations were conducted for both NEB barriers and stacking fault structures to find the underlying deformation mechanisms.
Chemically tailored planar fault defect phases in Ta-Fe µ-phases (manuscript in progress) Electron Backscatter Diffraction (EBSD) maps of randomly selected areas of Ta-Fe µ-phase samples at various compositions exhibit distinctive twinning characteristics. The twins observed in the first two µ-phase samples with 46 and 50 at.\% Ta exhibit only basal (0001) twins. As the Ta content of the Ta-Fe µ-phase increases, the orientation relationship and the twin plane change. In case of the µ-phase samples with 54 and 58 at.\% Ta, the pyramidal twin plane appears to be predominantly observed. To investigate the thermodynamic formation of twins, defect formation energy was calculated with respect to the pristine µ-phase.
Gasper, C., Soysal, E. M., Ulumuddin, N., Stollenwerk, T., Reclik, T., Sun, P. L., \& Korte-Kerzel, S. (2025). Mechanical properties and deformation mechanisms of the C14 Laves and µ-phase in the ternary Ta-Fe (-Al) system. Materials \& Design, 113625.
The Young’s modulus for different compositions in the Ta-Fe(-Al) system as calculated by DFT is plotted in parallel to experimentally measured indentation modulus in Figure 2. The elastic moduli of antiferromagnetic, ferromagnetic and ferrimagnetic TaFe2 Laves phases are shown to provide insight into the effect of magnetic configurations. Upon comparing the effect of composition within the binary µ-phase and ternary Laves phase, the lowest-energy magnetic configurations were antiferromagnetically-ordered Fe and ferromagnetically-ordered Fe, respectively.
Discussion
Across the presented studies, a unifying theme emerges: the mechanical behavior of structurally complex intermetallics is governed not only by their crystallographic building blocks but also by local bonding and defect chemistry. From the Sm–Co system, where deformation mechanisms evolve with structural complexity, to the Ta–Fe(–Al) phases, where composition and magnetic ordering subtly tune mechanical stiffness, these works collectively deepen the understanding of plasticity and defect energetics in intermetallics.
Advancing from empirical and atomistic modeling toward predictive, transferable structure–property relationships requires integrating ab initio energetics, interatomic potential development, and experimental validation. Future research will focus on investigating bonding changes during slip mechanisms. Such efforts will enable a more comprehensive description of non-crystallographic slip and phase-specific deformation, guiding the design of next-generation intermetallic materials.
Additional Project Information
DFG classification: 405-01 Metallurgical and Thermal Processes, Thermomechanical Treatment of Materials
Software: VASP
Cluster: CLAIX
Publications
Wei Luo, Christina Gasper, Siyuan Zhang, PL Sun, N Ulumuddin, A Petrova, Y Lysogorskiy, Ralf Drautz, Zhuocheng Xie, Sandra Korte-Kerzel.
Non-basal plasticity in the μ-phase at room temperature
Acta Materialia, 2025
Christina Gasper, Elif Merve Soysal, Nisa Ulumuddin, Tobias Stollenwerk, Tom Reclik, Pei-Ling Sun, Sandra Korte-Kerzel.
Mechanical properties and deformation mechanisms of the C14 Laves and µ-phase in the ternary Ta-Fe(-Al) system.
Materials & Design, 2025
Tobias Stollenwerk, Pia Carlotta Huckfeldt, Nisa Zakia Zahra Ulumuddin, Malik Schneider, Zhuocheng Xie, Sandra Korte‐Kerzel.
Beyond Fundamental Building Blocks: Plasticity in Structurally Complex Crystals.
https://dx.doi.org/10.1002/adma.202414376, 2025
Tobias Stollenwerk, Nisa Ulumuddin, Pei-Ling Sun, Sang-Hyeok Lee, Mattis Seehaus, Konstantin Skokov, Oliver Gutfleisch, Zhuocheng Xie, Sandra Korte-Kerzel
Dislocation-Mediated Plasticity in the Intermetallic Smco5 Phase
https://dx.doi.org/10.2139/ssrn.4600370, 2023