Project
Machine-learning Interatomic Potential for High Entropy Half Heusler Materials
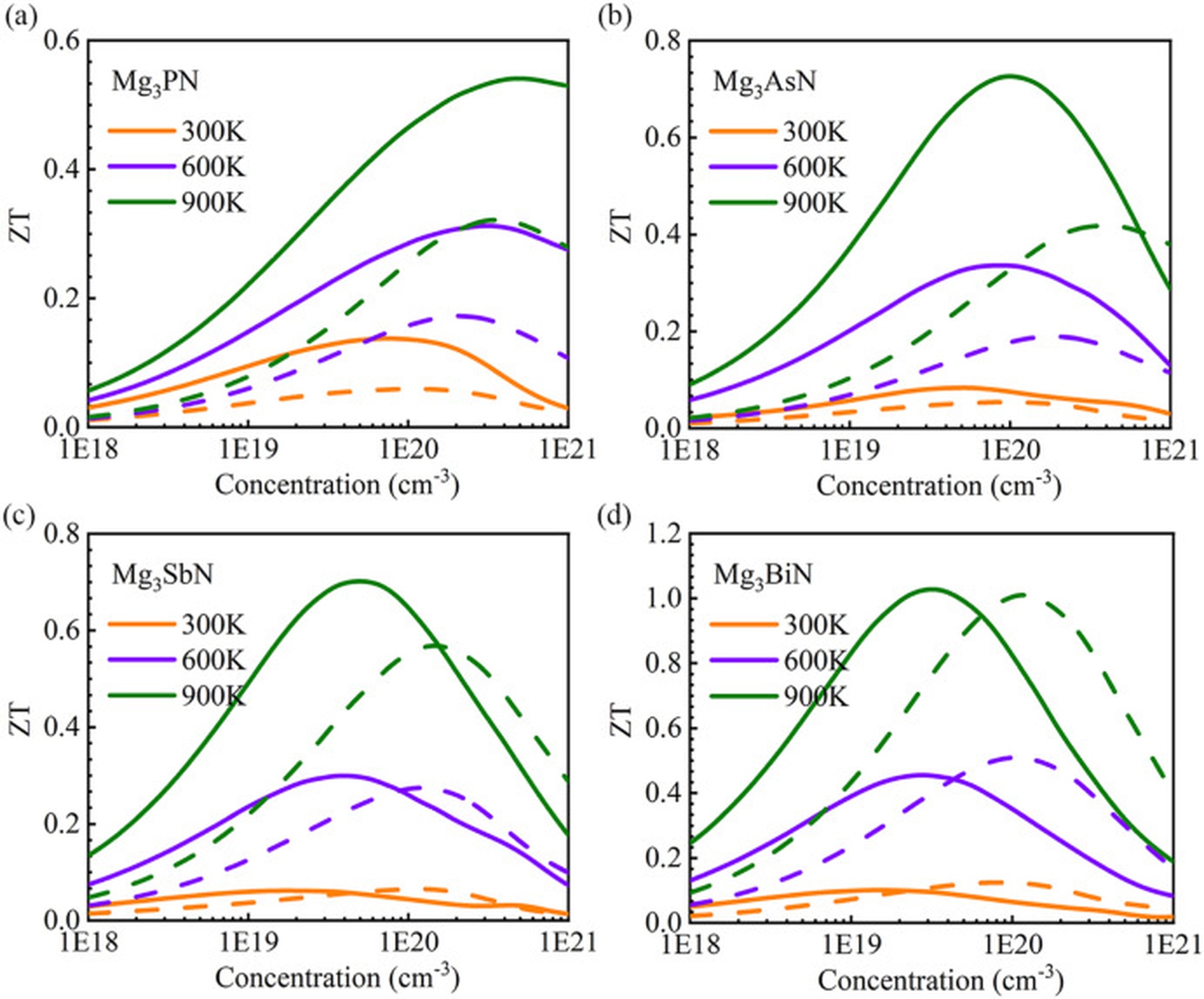 Figure 1: The thermoelectric figure of merit ZT as a function of carrier doping concentration of (a) Mg3PN, (b) Mg3AsN, (c) Mg3SbN, and (d) Mg3BiN at 300, 600, and 900 K
Figure 1: The thermoelectric figure of merit ZT as a function of carrier doping concentration of (a) Mg3PN, (b) Mg3AsN, (c) Mg3SbN, and (d) Mg3BiN at 300, 600, and 900 K 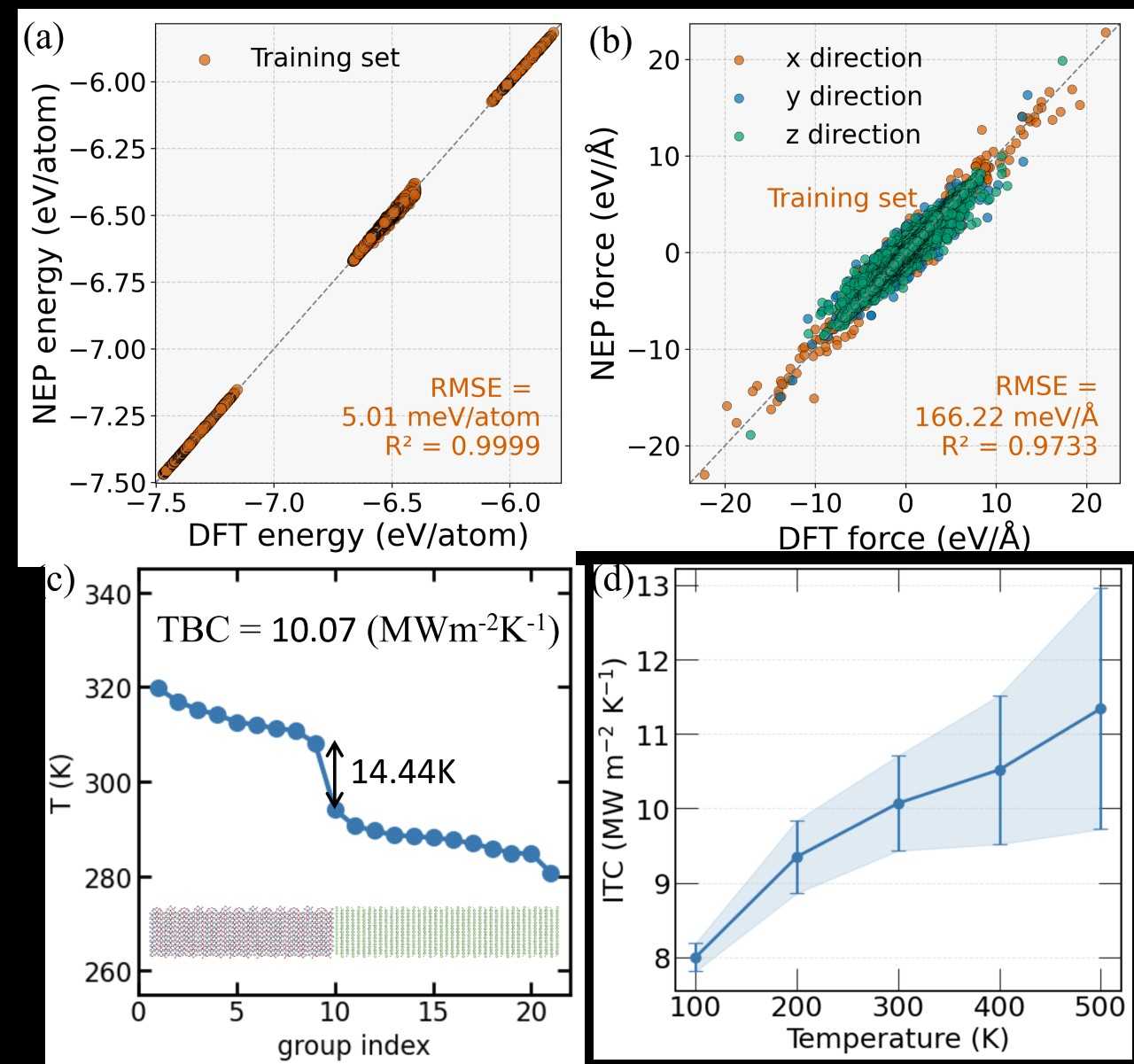 Figure 2: Comparison between DFT and NEP potentials for the interface: (a) energy and (b) forces. (c) Temperature profile of the interface. (d) Interfacial thermal conductance as a function of temperature.
Figure 2: Comparison between DFT and NEP potentials for the interface: (a) energy and (b) forces. (c) Temperature profile of the interface. (d) Interfacial thermal conductance as a function of temperature. 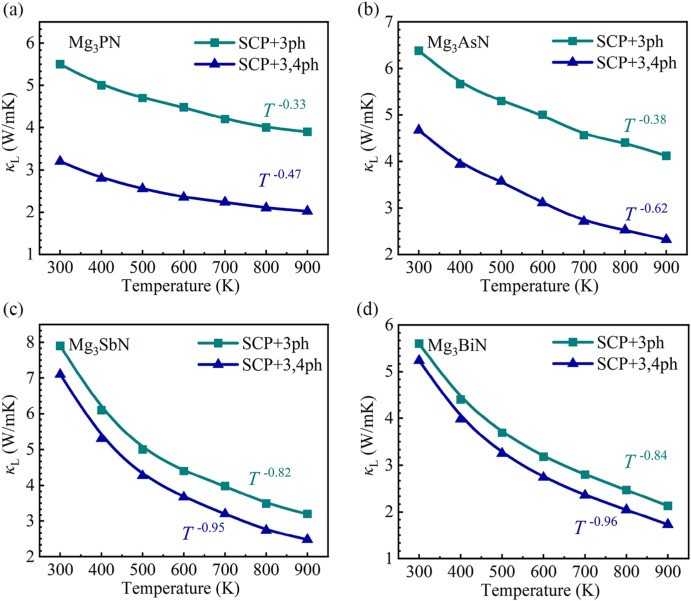 Figure 3: Calculated temperature-dependent thermal conductivity considering three-phonon and four-phonon processes.
Figure 3: Calculated temperature-dependent thermal conductivity considering three-phonon and four-phonon processes. Addressing the urgent challenges of the energy crisis and global warming has become imperative. Thermoelectric (TE) materials offer a promising route to convert heat into electrical energy without generating air pollution. High-throughput (HTP) calculations have emerged as an effective tool for screening potential TE candidates. Among these, antiperovskites have attracted considerable interest for applications in optoelectronics, thermoelectrics, transparent contacts, and thin-film transistors, yet many promising compounds remain unexplored. In this work, we combine HTP with first-principles calculations to evaluate the electronic and thermal transport properties of 4,281 compounds. Furthermore, motivated by the potential of interface systems in TE applications but their high computational cost, we implement a Machine Learning Interatomic Potential (MLIP) framework integrated with the Green–Kubo method to accelerate heat transport predictions, enabling a broader search for high-performance TE materials.
Project Details
Project term
July 1, 2024–June 30, 2025
Affiliations
TU Darmstadt
Institute
Theory of Magnetic Materials
Project Manager
Principal Investigator
Methods
Density functional theory (DFT) calculations were performed using VASP with a plane-wave basis and the PBEsol exchange–correlation functional. Electronic transport was evaluated using AMSET, with band gaps corrected via the HSE06 hybrid functional. Phonon band structures were computed with Phonopy using harmonic interatomic force constants from the finite-displacement method, and phonon dispersions obtained by diagonalizing the dynamical matrix. Thermal transport was calculated via the Boltzmann Transport Equation (BTE), Equilibrium Molecular Dynamics (EMD), and Non-Equilibrium Molecular Dynamics (NEMD).
For MLIP training, datasets combined MD simulations and static DFT calculations, with forces, energies, and stresses serving as reference data.
Results
1. Thermoelectric properties of antiperovskite
We investigated cubic APV materials A3XN (A = Sr, Mg, Ca; X = P, As, Sb, Bi) using advanced anharmonic phonon theory and electron–phonon coupling. Our results reveal that strong quartic anharmonicity plays a decisive role in reducing lattice thermal conductivity and modifying its temperature dependence, deviating from the conventional inverse-temperature trend.
In Sr-based compounds, dispersive band edges and band degeneracy lead to enhanced electrical transport properties. In particular, Sr-containing bismuth nitrides stand out with superior thermoelectric performance.
Mg-based APVs exhibit diverse behaviors: some compounds show strong phonon anharmonicity, while others feature localized vibrational modes that suppress heat transport. Both effects contribute to favorable thermoelectric performance at elevated temperatures.
Ca-based APVs display pronounced anharmonicity and particularly low lattice thermal conductivity. Several of these compounds maintain good thermoelectric efficiency across a broad temperature range, identifying them as promising candidates for practical applications.
2. Heat transport for interface
For the Al2O3–GaN interface, a machine-learning interatomic potential (MLIP) was developed and validated against first-principles calculations. The MLIP reproduces phonon spectra and thermal conductivities of the constituent materials with high accuracy.
Simulations of interfacial heat transport reproduce experimental observations and reveal that thermal boundary conductance depends strongly on atomic termination. Interfaces terminated with different atomic species display distinct heat transfer characteristics, highlighting the role of interfacial chemistry in phonon scattering and transmission.
Discussion
Our study demonstrates that intrinsic anharmonicity is a key factor in suppressing lattice thermal conductivity in APV materials, thereby enabling high thermoelectric performance. Compounds that combine strong anharmonic effects with favorable electronic band structures achieve promising efficiency, with several members identified as high-ZT candidates even at medium operating temperatures.
For oxide–nitride interfaces such as Al2O3–GaN, machine-learning interatomic potentials provide an accurate and efficient route to model interfacial transport. The sensitivity of thermal boundary conductance to atomic termination suggests practical pathways to engineer heat flow by tuning interfacial bonding and reconstruction.
Together, these findings highlight the dual importance of anharmonic phonon engineering in bulk materials and interface design in heterostructures. By integrating high-throughput screening, advanced phonon theory, and MLIPs, we establish a general framework for designing efficient thermoelectric materials and low-resistance interfaces for energy and electronic applications.
Additional Project Information
DFG classification: 406 Materials Science
Software: VASP, Phonopy, AMSET, ShengBTE, GPUMD
Cluster: Lichtenberg
Publications
[1] Yue, J.; Liu, Y.; Ren, W.; Lin, S.; Shen, C.; Singh, H. K.; Cui, T.; Tadano, T.; Zhang, H. Role of atypical temperature-responsive lattice thermal transport on the thermoelectric properties of antiperovskites Mg₃XN (X = P, As, Sb, Bi). Materials Today Physics (2024). https://doi.org/10.1016/j.mtphys.2024.101340
[2] Lin, S.; Yue, J.; Ren, W.; Shen, C.; Zhang, H. Strong anharmonicity and medium-temperature thermoelectric efficiency in antiperovskite Ca₃XN (X = P, As, Sb, Bi) compounds. Journal of Materials Chemistry A (2024), 12, 19567–19579 https://doi.org/10.1039/D4TA02118E