Project
Computational design of nanopores
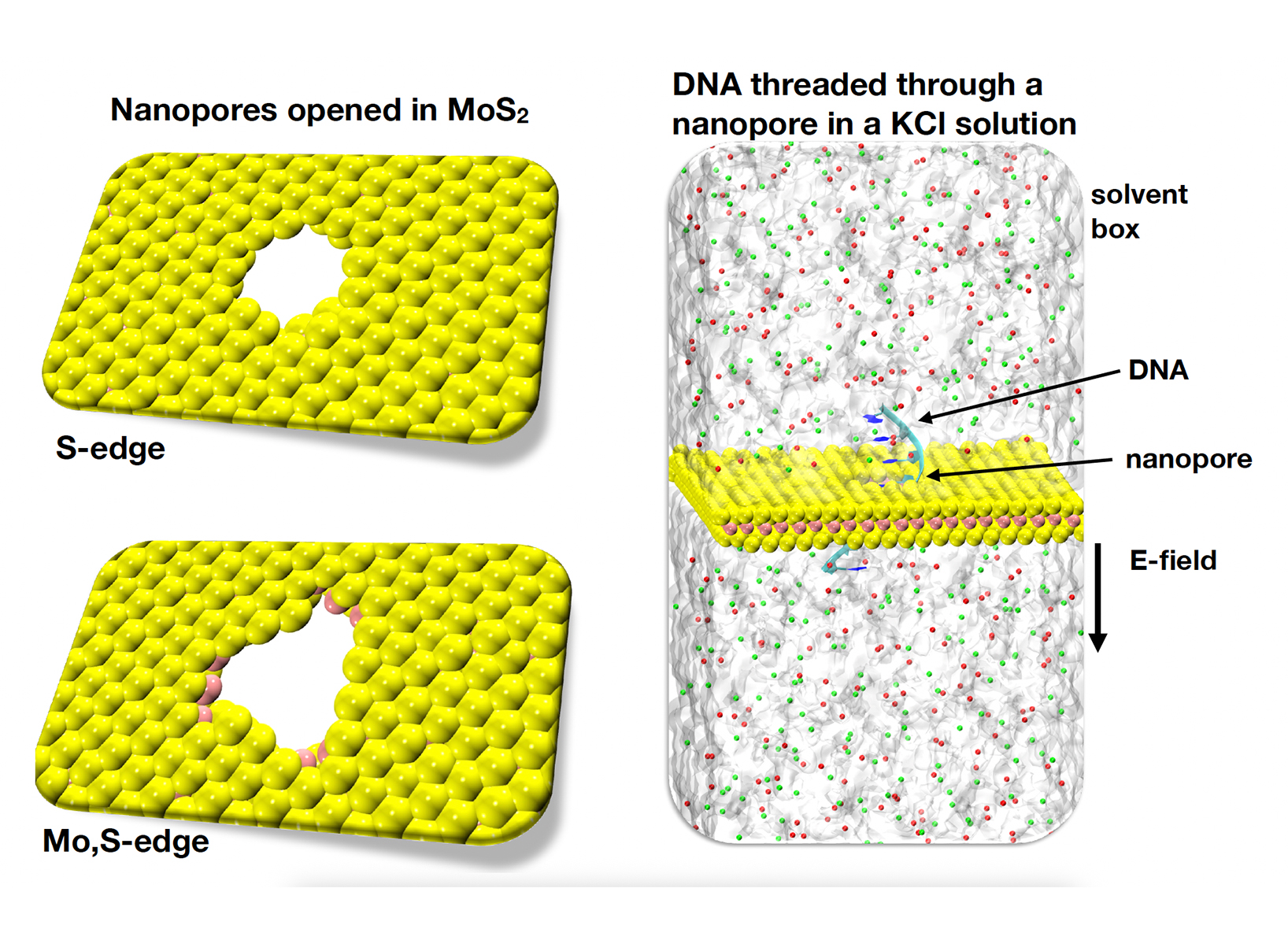 Figure 1: Left: the top views of an S-edge (top) and a mixed Mo,S-edge (bottom) MoS2 nanopore, respectively. The S and Mo atoms are colored pink and yellow, respectively. The opening of the pores is about 1.5 nm in diameter. Right: the nanopore setup showing a DNA molecule threading the pore within a KCl salt solution under the application of an external electric field (E), as denoted by the legends. The water molecules are represented by the light gray background, while the K+ and Cl− ions are colored in green and red, respectively.
Figure 1: Left: the top views of an S-edge (top) and a mixed Mo,S-edge (bottom) MoS2 nanopore, respectively. The S and Mo atoms are colored pink and yellow, respectively. The opening of the pores is about 1.5 nm in diameter. Right: the nanopore setup showing a DNA molecule threading the pore within a KCl salt solution under the application of an external electric field (E), as denoted by the legends. The water molecules are represented by the light gray background, while the K+ and Cl− ions are colored in green and red, respectively.  Figure 2: The top and side views of (a) Fe-Pc and (b) OFe-Pc, respectively. The color codes are given in the l.
Figure 2: The top and side views of (a) Fe-Pc and (b) OFe-Pc, respectively. The color codes are given in the l. 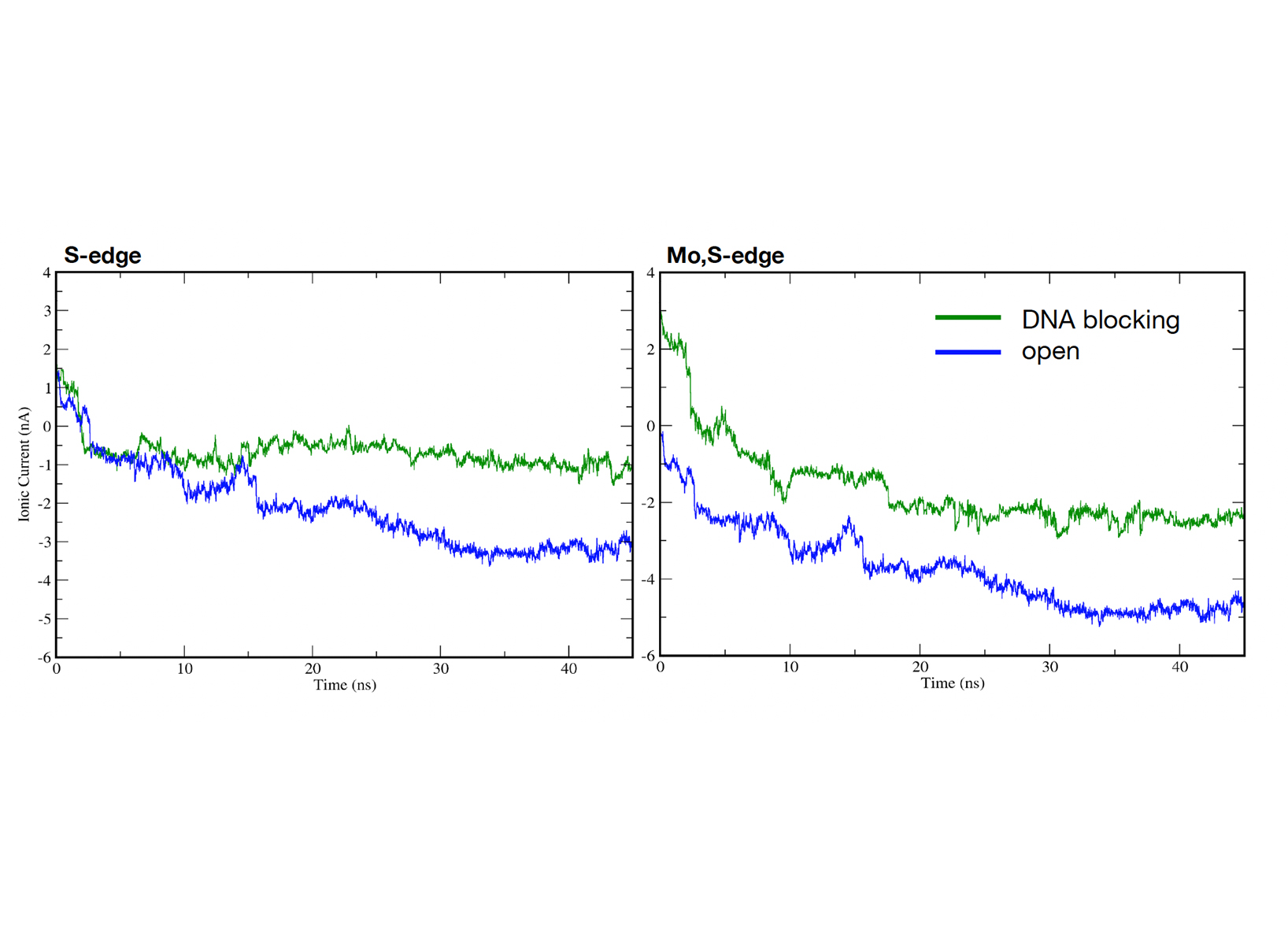 Figure 3: The dynamics of the ionic current blockades for (a) the S-edge and (b) the Mo,S-edge MoS2 nanopores. The results in each case are shown for both the open (no DNA passes the pore) and the blocked (with a DNA).
Figure 3: The dynamics of the ionic current blockades for (a) the S-edge and (b) the Mo,S-edge MoS2 nanopores. The results in each case are shown for both the open (no DNA passes the pore) and the blocked (with a DNA). 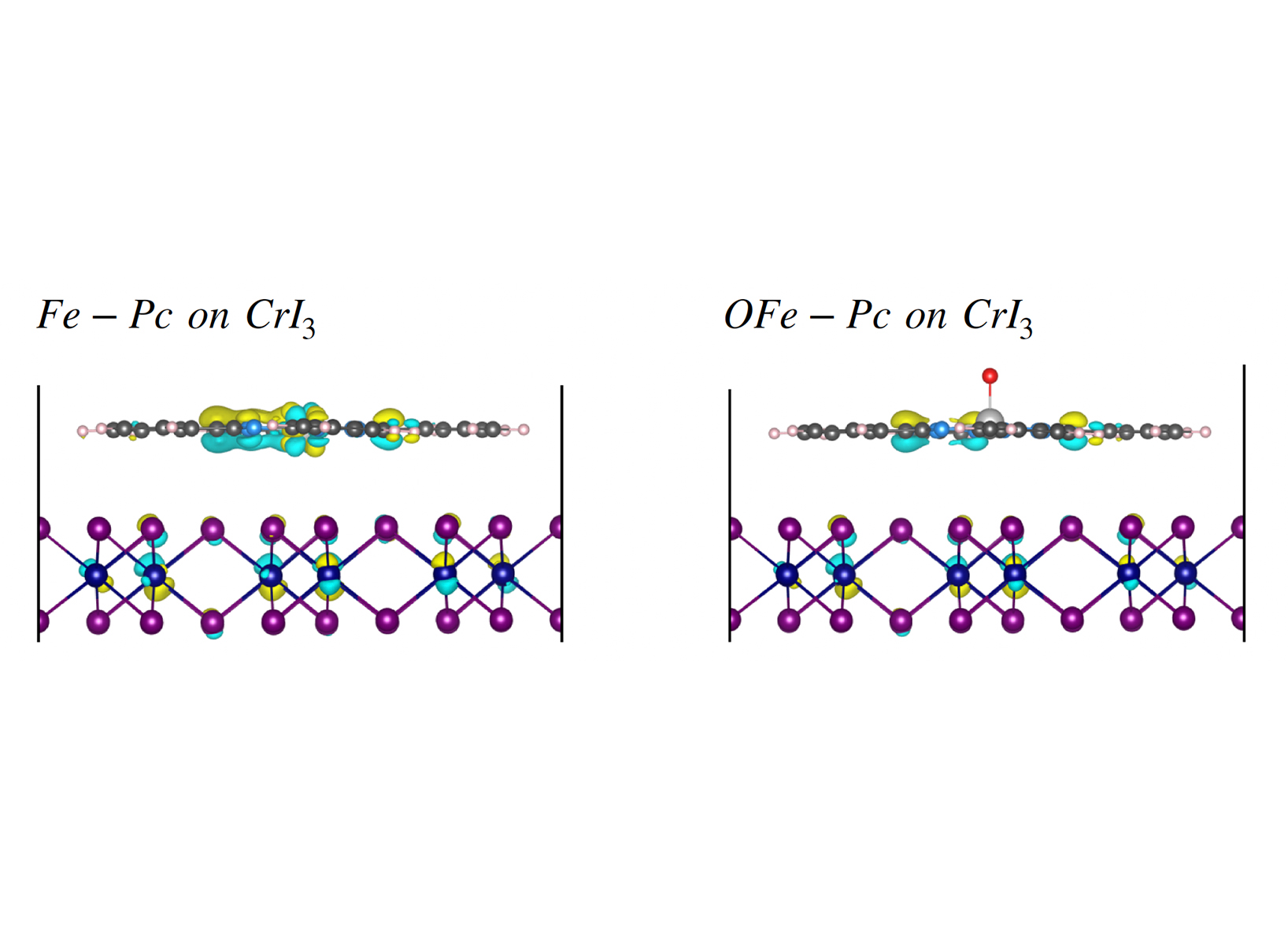 Figure 4: Side view of the charge density differences (in yellow and cyan) for the adsorbed Fe-Pc on CrI3, without (left) and with (right) oxygen. The color coding of the atoms follows that of Fig.2. The charge densities of pristine Fe-Pc (OFe-Pc) and CrI3 are subtracted from the charge density of the combined system representing the absorbed molecule on CrI3.
Figure 4: Side view of the charge density differences (in yellow and cyan) for the adsorbed Fe-Pc on CrI3, without (left) and with (right) oxygen. The color coding of the atoms follows that of Fig.2. The charge densities of pristine Fe-Pc (OFe-Pc) and CrI3 are subtracted from the charge density of the combined system representing the absorbed molecule on CrI3.  Figure 5: On-going quantum-mechanical simulations (using the code VASP) of a small nanopore opened in a 2D graphene sheet. The nanopore edge is terminated by hydrogen atoms (left). Different amino-acids named through the legends are separately being placed in the nanopore (right) in order to reveal aminoacids dependent signatures in the nanopore properties. The hydrogen, carbon, oxygen, nitrogen atoms are colored in white, gray, red, and blue, respectively.
Figure 5: On-going quantum-mechanical simulations (using the code VASP) of a small nanopore opened in a 2D graphene sheet. The nanopore edge is terminated by hydrogen atoms (left). Different amino-acids named through the legends are separately being placed in the nanopore (right) in order to reveal aminoacids dependent signatures in the nanopore properties. The hydrogen, carbon, oxygen, nitrogen atoms are colored in white, gray, red, and blue, respectively. This project focuses on the computational modeling of nanopores. These are nanometer-sized pores in materials that can electrophoretically thread biomolecules, such as DNA, RNA, and proteins. That is, an electric field applied across the nanopore opening can thread the biomolecules in the presence of a salt solution, water and ions. In this project, we have planned to model the nanopores as materials, as well as the threading of biomolecules, mainly DNA and short peptides. Such a modeling involves different time and length scales and needs to involve a variety of computational methods. A materials, representative of nanopores, we focus on two-dimensional (2D) structures, such as graphene (G) and molybdenum disulfide (Mo2) in which atoms are removed to form a nanopore opening. These are placed in a solution and DNA molecules are pulled through these. On top of these calculations, we additionally calculate the properties of another class of 2D materials, namely chromium iodine (CrI3). In this case, we are interested in its magnetic properties and how these are modified and can be tuned by adsorbing molecules on it. Overall, in both the fields of nanopores, as well as that of 2D materials (for electronics, nanopores and other applications) intense research is being carried on in order to (a) improve the biological sensing of nanopores and extend this to the detection of proteins and (b) optimize the setup and use of 2D materials for this purpose. Hand in hand with this aspects is the need to find or predict new 2D materials and unravel their properties in order to assess their potential use in nanopore sensing.
Project Details
Project term
May 1, 2022–April 30, 2023
Affiliations
RWTH Aachen University
Institute
Computational Biotechnology
Project Manager
Methods
In order to unravel the properties of the nanopore materials, as well as their sensing potential in view of detecting biomolecules, their length, identity, and sequence, simulation methods at the density functional theory (DFT) and the atomistic Molecular Dynamics (MD) level are performed. The DFT simulations have been performed for calculating the properties of two-dimensional materials, while MD was carried out for monitoring the dynamics of DNA through the 2D structures. Specifically for the simulations of the magnetic properties of CrI3 only DFT simulations were carried out. In order to clarify, which systems and cases were modelled in the reporting period, we summarize these in the following:
• MoS2 nanopore of about 1.5 nm in diameter placed in a potassium chloride (KCl) solution of 1M. A short DNA strand was pulled through the pore within the solution and the corresponding dynamics and ionic current traces through the pore are monitored. We name this ’pore system’. For this purpose, we have opened a small pore in a 2D MoS2 material by removing Mo and S atoms. This approach led to the formation of two nanopores with different edges: one with only S atoms at the nanopore edge (S-edge pore) and one with a nanopore edge with both Mo and S atoms (Mo,S-edge pore). These are depicted in Fig.1.
• An iron-phthalocyanines (Fe-Pc) molecule is placed on top of a 2D CrI3 material and the modification in the magnetic properties of the original pristine CrI3 structure is evaluated. For this a detailed analysis of the structure, as well the electronic behavior of the 2D material and the molecule on top of the 2D material is needed. We name the latter ’magnetic system’. Specifically, we have studied the oxygen-capture and -release properties of Fe-Pc on top of magnetic monolayer CrI3. For this, we have have simulated the adsorption of the Fe-Pc without (Fig.2(a)) and with (Fig.2(b)) an oxygen attached on the iron (Fe) atom of the Fe-Pc molecule (OFe-Pc), which is in turn adsorbed on the CrI3 structure (not shown in Fig.2).
Results
Representative incremental results on both systems listed above are discussed next.
From our simulations we could get insight on the following:
pore system An important goal of the simulations is the calculation of the ionic current through the pore, as a function of time. The ionic current is the quantity that is experimentally measured and can be directly compared to
the modeling results. For the pores depicted in Fig.1 the time series, i.e. the dynamics, of the ionic current through the pore are shown in Fig.3. As a first observation, the traces in the ionic current are different for the nanopores of different edges, revealing the influence of the exact pore edge chemistry in the ionic transport through the pore. In addition, the fact that the DNA molecule is bulkier than the ions and water in the solution and blocks more the pore region is mapped through the deeper ionic traces in this case.
magnetic system We have shed light into the modification of the monolayer CrI3 the Fe-Pc with and without the oxygen on the O atom Fig.4). This modification was directly mapped on the electronic properties of the combined Fe-Pc (with/without O) on the 2D CrI3 material. The energy minimization calculations resulted in the structures shown on Fig.4. The corresponding charge density differences is also shown. This was calculated as the addition of the charge densities of individual CrI3 and Fe-Pc is subtracted from the charge density of Fe-Pc/CrI3. The electrons deplete from the green regions to yellow regions while Fe-Pc/CrI3 is formed (see Fig.4). The closer look to Fe reveals that the interaction between molecule and the layer changes the orbital occupancy of Fe atoms. When the oxygen atom (O)is attached on the iron atom (Fe) forming the OFe-Pc molecule, no charge difference is formed in the vicinity of Fe atom.
Discussion
Again listed with respect to the two different systems, we discuss briefly the most
important insights that our simulations could deliver, together with a short outlook.
pore system We have modelled nanopore materials made of MoS2 and placed these in a salt solution in order to monitor the translocation (passage) of DNA molecules through. This passage is being evaluated by the ionic current traces through the nanopores and can be compared with the experimental measurements. This part of our work focused on the influence if the pore chemistry on these traces. The long-term goal of our simulations aims to optimize the readout of different biomolecules through different nanopore setups. The latter involve various materials including MoS2, as well as graphene and hexagonal boron-nitride and their combinations. On top of detecting DNA and its sequence, we also turn to the detection of short peptides threading these nanopores. Our most recent simulations within the framework of the continuation of this computing project, indeed also involve graphene nanopores translocated by peptides of various amino-acid sequences. Snapshots of our first simulations along these lines are depicted in Fig.5. The contribution of our simulations in the field of nanopore sensing also involves the optimization of the conditions, such as the salt concentration, the electric field the use of linker molecules that drag the peptides to the pore, etc. A very specific aspect in nanopore sequencers is the nanopore chemistry, as well as the nanopore setup, along which we are also currently performing simulations.
magnetic system The interplay between the magnetic properties of Fe-Pc on CrI3 and its chemical activity are investigated. We found that the surface effects on the molecule accompanied with the magnetic interactions between Fe and Cr atoms can be used to manipulate – even control – the oxygen capturerelease properties of Fe-Pc. We have also unravelled the influence of defects on the 2D material on these properties. This work is the basis of followup simulations on the adsorption of other types of molecules, such as DNA units (nucleobases) and/or protein units (amino-acids) on other types of 2D materials, such as Mo2, graphene, and hexagonal boron-nitride.
Additional Project Information
DFG classification: 201-02 Biophysics, 302-03 Chemical Solid State and Surface Research, Theory and Modelling, 307 Condensed Matter Physics
Software: VASP, Gromacs
Cluster: CLAIX
Publications
C. Bacaksiz and M. Fyta, Fe-Pc molecule on top of monolayer CrI3: a controllable
platform for oxygen capturing and releasing by Fe-Pc (in preparation).