Project
First Principles Ab Initio Thermodynamics of Single Atom Alloy Catalysts
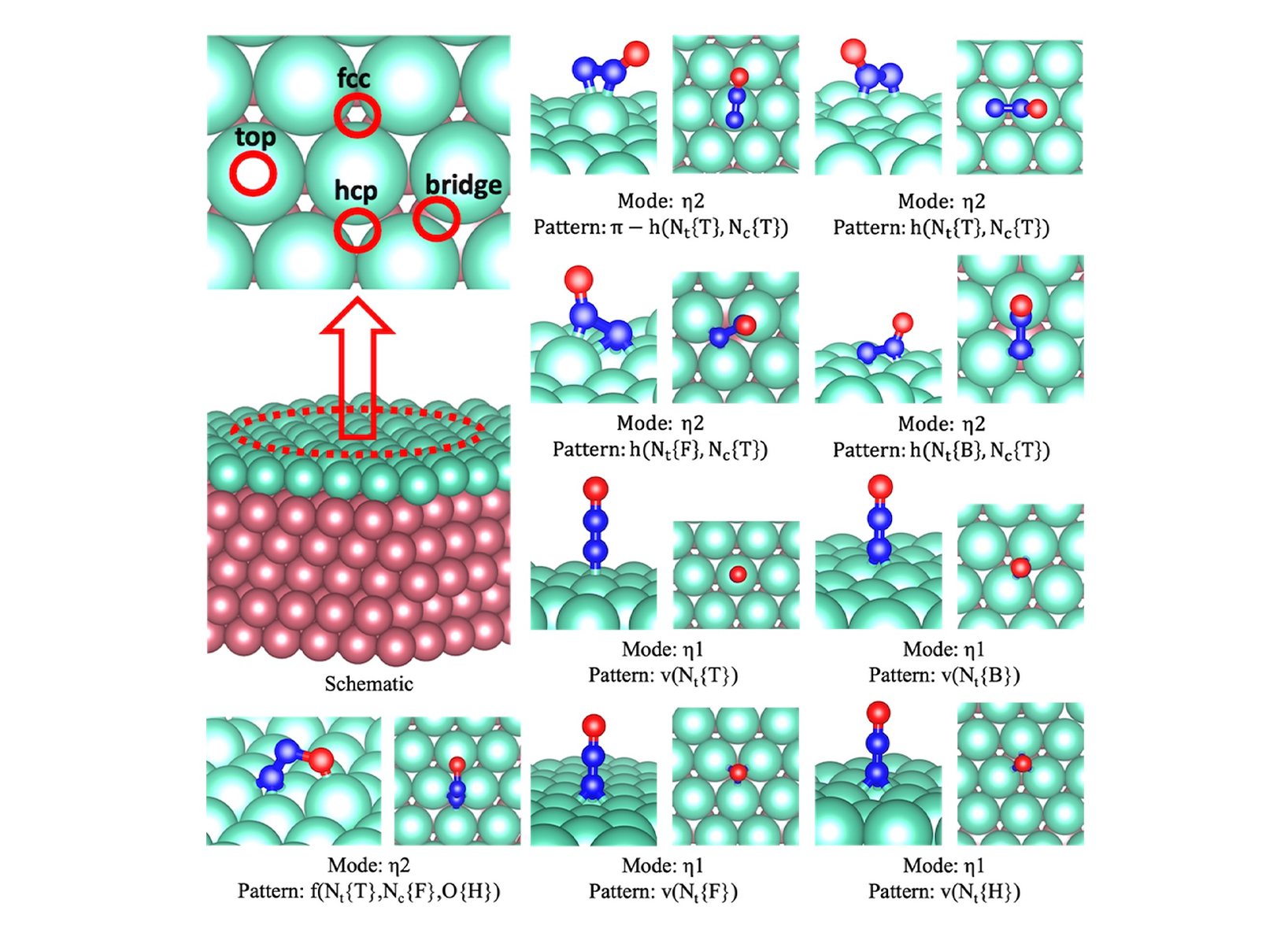 Some examples of N2O adsorbate configurations obtained from DFT calculations shown in side and top views (from results section 3.2)
Some examples of N2O adsorbate configurations obtained from DFT calculations shown in side and top views (from results section 3.2) 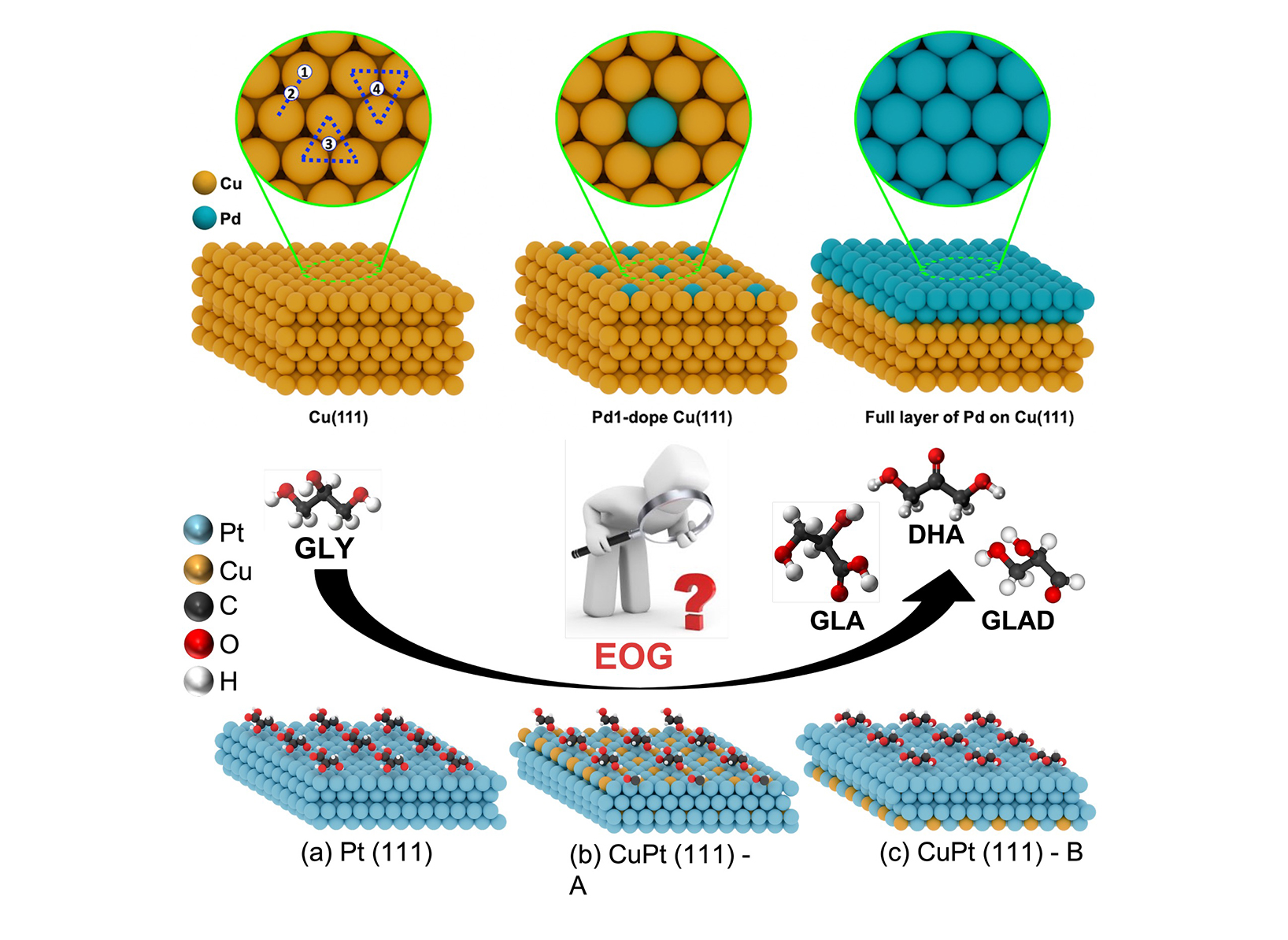 (top) Atomistic view of slab models of SAA Catalsys (from results section 3.3) (bottom) Schematic representation of catalyst surface models with adsorbed intermediates (from results section 3.4)
(top) Atomistic view of slab models of SAA Catalsys (from results section 3.3) (bottom) Schematic representation of catalyst surface models with adsorbed intermediates (from results section 3.4) Catalysts are critical to many industrial processes, especially for reducing harmful emissions in automotive and energy applications. Single-Atom Alloy (SAA) catalysts have shown great promise in this area. These materials involve placing individual atoms of expensive, high-performance metals (like Pt, Rh, or Ru) onto/into the surface of cheaper base metals (such as Cu or Ni). This not only reduces the amount of costly metals needed but can also enhance the catalyst’s efficiency and selectivity. However, the success of such catalysts depends heavily on the precise arrangement of atoms at the surface, known as the surface morphology. Predicting how these catalysts behave at various temperatures and concentrations is a complex task that cannot be solved by simple trial and error. It requires understanding interactions at the atomic scale across thousands of possible configurations. This is where configurational sampling becomes essential and instrumental.
Project Details
Project term
March 15, 2024–June 6, 2025
Affiliations
RWTH Aachen University
Institute
The Fuel Science Center
Principal Investigator
Methods
Our approach involved combining multiple levels of simulation and data modeling to strike a balance between accuracy and computational cost. We began by generating a wide range of atomic structures using a tool CASM that creates all possible ways atoms can be arranged on a metal surface. These included different concentrations and placements of noble metal atoms on the surface of copper and nickel.
To quickly evaluate these structures, we first used an approximate but fast GFN-xTB method to estimate their relative stability. From this pool, we selected the most promising candidates and refined their properties using highly accurate physical simulations using VASP software. These high-accuracy simulations are computationally expensive, so we limited them to only the most relevant structures. To go further, we developed a MACE ML model based on the known results. This ML model could estimate the energy of new structures without having to simulate each one in detail. Using this model, we performed Monte Carlo (MC) simulations that mimic how atoms move and arrange themselves at different temperatures, something crucial for understanding the real-world behavior of catalysts.
Results
Over the course of the project, the research evolved through four distinct but interconnected sub-project, as discussed below.
3.1 MC simulations for dopant distribution in SAA catalyst surfaces
We applied the computational workflow to SAA systems (Cu-Pd, Cu-Pt, Cu-Rh, Cu-Ru, Ni-Rh, Ni-Ru), screening over 6,000 atomic configurations with fast approximations and refining 2,000 via DFT. We trained a MACE machine-learned potential on DFT data for large-scale MC simulations to study temperature- and composition-dependent surface morphologies. Preliminary results showed isolated noble metal atoms dominate at low concentrations, favoring activity, while higher concentrations lead to clustering or subsurface migration, reducing active sites. This study is still ongoing.
3.2 Mechanistic insights into N₂O decomposition on hybrid catalyst surfaces within the framework of SAA design
This study investigated N₂O decomposition on hybrid catalysts of precious (Ru, Rh) and non-precious (Cu, Ni) metals, and their alloys, with particular focus on the influence of surface-adsorbed NO. The results showed that NO suppresses activity by modifying the adsorption behavior of key intermediates. Hybrid systems, especially Ru-Ni and Ru-Cu alloys, exhibited improved performance, with Ni-based compositions maintaining higher activity even in presence of NO. Notably, conventional activity descriptors reversed in the presence of NO, highlighting that designs ignoring co-adsorbate effects may mispredict realistic catalytic performance. This highlights the importance of accounting for co-adsorbate effects, metal combinations, and their interactions with intermediates when designing SAA catalysts for practical applications.
3.3 SAA catalysts for H₂-selective catalytic reduction
Hydrogen dissociation is a critical step in many catalytic processes. Collaborating with Prof. Regina Palkovits’ experimental group at ITMC, we studied Pd-doped Cu(111) SAA to design an efficient catalyst that maximize activity while minimizing precious metal content. DFT and CI-NEB calculations showed that a single Pd atom lowers the H₂ dissociation barrier from 0.46 eV (pure Cu) to 0.245 eV, confirming its catalytic role. While higher Pd concentrations further reduce the barrier, isolated Pd sites already enhance activity significantly. These results are being prepared for publication.
3.4 SAA Catalysts for Glycerol Oxidation
In another ongoing collaboration with ITMC, we explored a Cu-Pt SAA catalyst for glycerol electro-oxidation, a key step in sustainable biomass valorization. DFT results showed that pure Pt(111) needs high electrode potentials for GLAD formation versus DHA, while alloying with Cu modifies surface properties, lowering required potentials and shifting selectivity toward GLAD. The results are being prepared for publication.
Discussion
MC simulations revealed valuable insights into dopants distribution on catalyst surfaces across different temperatures and concentrations, favoring isolated atoms at low concentration, and clustering or migration at higher ones. However, for accurate prediction of stable configurations, these MC simulations must be coupled with atomic-level optimization that ensure precise energy evaluation alongside broad configurational sampling. This foundational understanding of dopant behavior sets the stage for interpreting catalytic activity trends across different alloy systems. For instance, we observed a breakdown of conventional scaling relations, particularly in the presence of co-adsorbed NO in N2O decomposition work. This deviation is more pronounced in SAAs than in hybrid systems, where isolated active sites fundamentally shift surface chemistry, opening new avenues for catalyst design beyond conventional activity limits. Similar effects were seen in hydrogen dissociation and glycerol oxidation, where alloying enhanced reactivity and selectivity. Future work should integrate these methods with dynamic adsorbate effects under operando conditions for improved SAA catalyst design.
Additional Project Information
DFG classification: 406-01 Thermodynamics and Kinetics of Materials
Software: VASP, LAMMPS, CASM, GFN-xTB
Cluster: CLAIX
Publications
1. Publication on SAA Catalysts for Glycerol Oxidation expected to be published by end of the year 2025
2. Publication on SAA catalysts for H₂-selective catalytic reduction expected to be published by end of the year 2025
3. Masters thesis on Monte Carlo Simulations of SAA catalysts for various guest host combinations to be finished by end of September 2025